

Biochemical and molecular analysis in mucopolysaccharidoses: what a paediatrician must know
- PMID: 30442161
- PMCID: PMC6238298
- DOI: 10.1186/s13052-018-0553-2
Biochemical and molecular analysis in mucopolysaccharidoses: what a paediatrician must know
Abstract
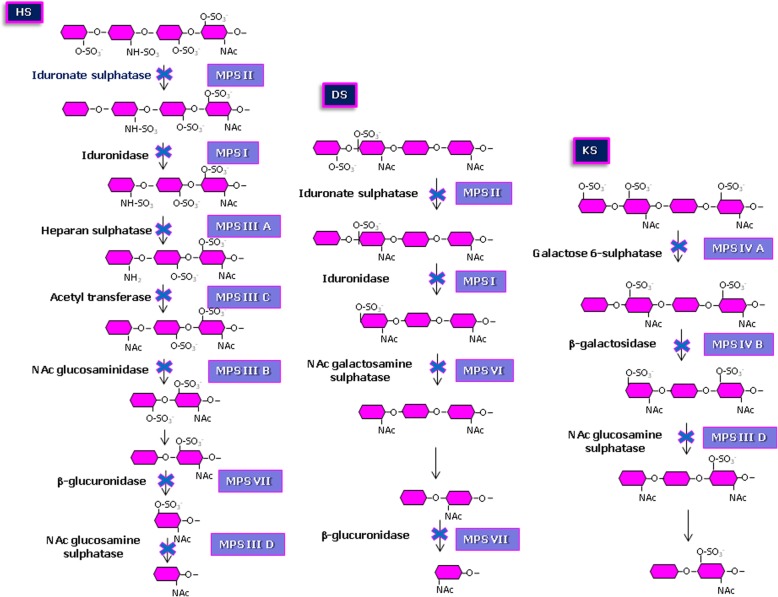
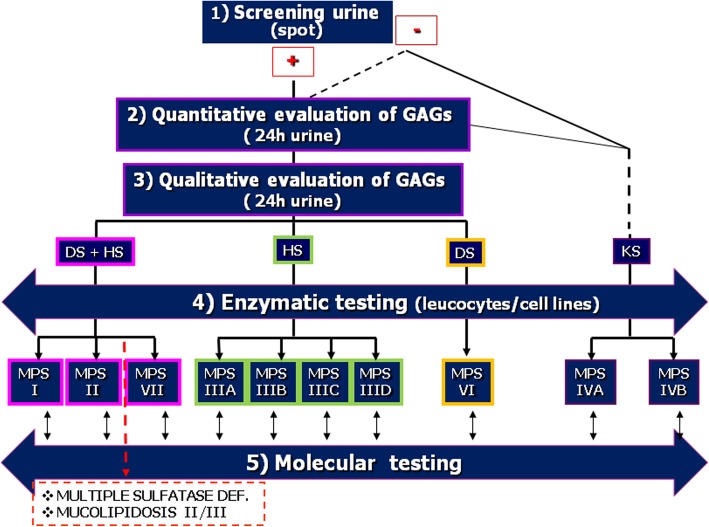
Mucopolysaccharidoses (MPS) are rare inherited disorders caused by a deficit of the lysosomal hydrolases involved in the degradation of mucopolysaccharides, also known as glycosaminoglycans (GAGs). They are all monogenic defects, transmitted in an autosomal recessive way, except for MPS type II which is X-linked. The enzymatic deficit causes a pathologic accumulation of undegraded or partially degraded substrates inside lysosomes as well as in the extracellular compartment. MPS generally present with recognizable signs and symptoms to raise a clinical suspicion. However, although they have individual peculiarities, often signs and symptoms may overlap between different MPS types. Therefore, a deeper evaluation of specific disease biomarkers becomes necessary to reach an appropriate diagnosis. This paper stresses the central role of the laboratory in completing and confirming the clinical suspicion of MPS according to a standardized procedure: first, a biochemical evaluation of the patient samples, including qualitative/quantitative urinary GAG analysis and a determination of enzyme activities, and then the molecular diagnosis. We also encourage a constant and close communication between clinicians and laboratory personnel to address a correct and early MPS diagnosis.
Keywords: Genetic counselling; Genotype-phenotype relationship; Glycosaminoglycans; Laboratory tests; Lysosomal storage disorders; Molecular analysis; Mucopolysaccharides; Pseudodeficiency.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Coppa GV, Catassi C, Gabrielli O, Giorgi PL, Dall'Amico R, Naia S, et al. Clinical application of a new simple method for the identification of mucopolysaccharidoses. HelvPaediatr Acta. 1987;42:419–423. - PubMed
-
- Wijburg FA, Węgrzyn G, Burton BK, Tylki-Szymańska A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013;102:462–470. doi: 10.1111/apa.12169. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
