

A new case report of severe mucopolysaccharidosis type VII: diagnosis, treatment with haematopoietic cell transplantation and prenatal diagnosis in a second pregnancy
- PMID: 30442200
- PMCID: PMC6238262
- DOI: 10.1186/s13052-018-0566-x
A new case report of severe mucopolysaccharidosis type VII: diagnosis, treatment with haematopoietic cell transplantation and prenatal diagnosis in a second pregnancy
Abstract
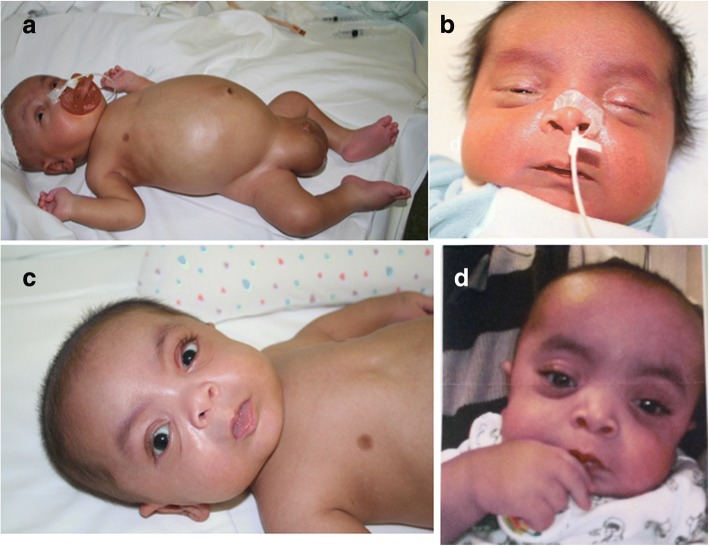
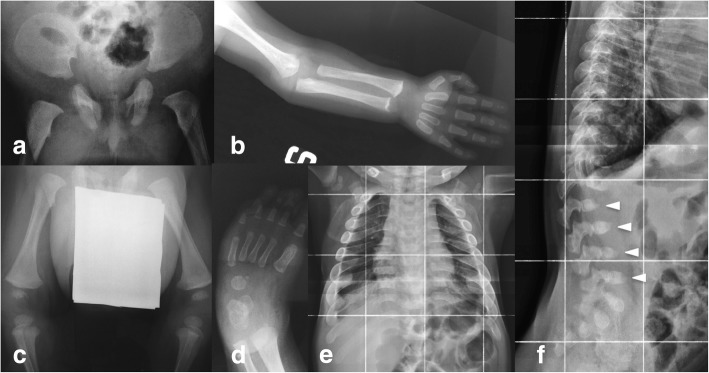
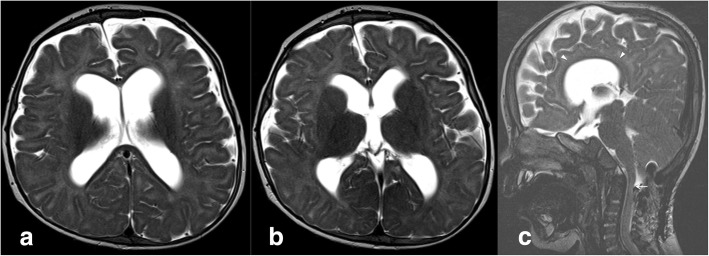
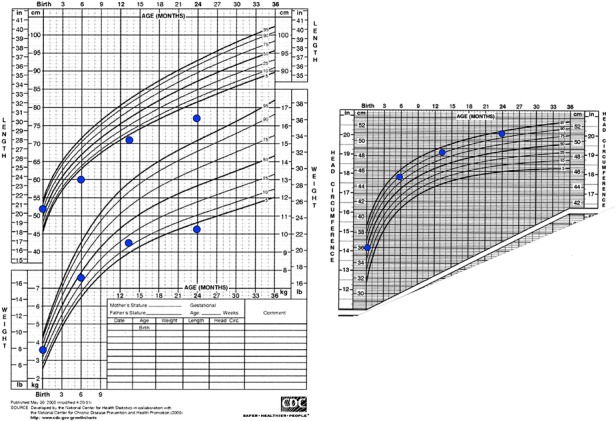
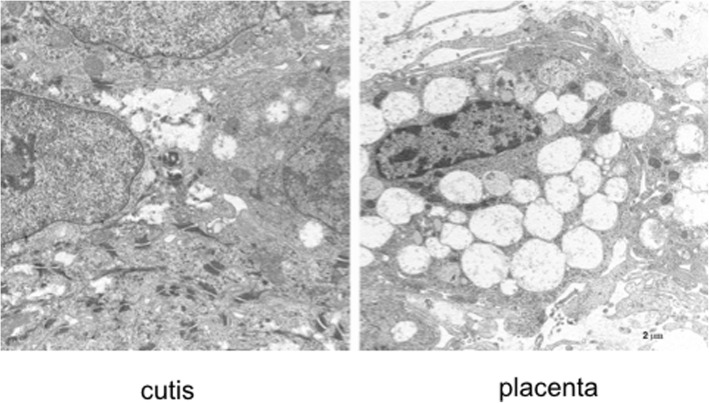
A new patient with severe mucopolysaccharidosis (MPS) type VII is reported. Non-immune hydrops fetalis (NIHF) was diagnosed during pregnancy. At birth, he showed generalized hydrops and dysmorphic features typical of MPS. Many diagnoses were excluded before reaching the diagnosis of MPS VII at 8 months of life. During the first year of life he had frequent respiratory infections associated with restrictive and obstructive bronchopneumopathy and underwent three surgical interventions: decompression of the spinal cord at the craniocervical junction, bilateral inguinal hernia, and bilateral clubfoot. At 14 months of life he underwent successful haematopoietic cell transplantation (HCT). During the following 10 months, his bronchopneumopathy progressively worsened, needing chronic pharmacological treatment and O2 administration. The patient died of respiratory insufficiency during a respiratory syncytial virus infection at 25 months of age. Molecular analysis showed the homozygous variant c.1617C > T, leading to the synonymous mutation p.Ser539=. This caused aberrant splicing with partial skipping of exon 10 (r.1616_1653del38) and complete skipping of exon 9 (r.1392_1476del85; r.1616_1653del38). No transcript of normal size was evident. The parents were both confirmed to be carriers. In a subsequent pregnancy, a prenatal diagnosis showed an affected fetus. Ultrasound examination before abortion showed NIHF. The skin and placenta examination by electron microscopy showed foamy intracytoplasmic vacuoles with a weakly electron-dense substrate. MPS VII is a very rare disease but it is possible that some cases go undiagnosed for several reasons, including that MPS VII, and other lysosomal storage diseases, are not included in the work-up for NIHF in many institutions, and the presence of anasarca at birth may be confounding for the recognition of the typical facial characteristics of the disease. This is the eighth patient affected by MPS VII who has undergone HCT. It is not possible to draw conclusions about the efficacy of HCT in MPS VII. Treatment with enzyme replacement is now available and will probably be beneficial for the patients who have a milder form with no or little cognitive involvement. Increased awareness among clinicians is needed for prompt diagnosis and to offer the correct treatment as early as possible.
Keywords: Beta-glucuronidase; GUSB gene; HCT; Haematopoietic cell transplantation; LSDs; MPS VII; Mucopolysaccharidosis; NIHF; Non-immune hydrops fetalis.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
The family consented that the data and images of the child were published in this paper.
Competing interests
Rossella Parini received a honorarium for participation in an Advisory Board meeting supported by Ultragenyx. The other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation.Int J Mol Sci. 2019 Oct 28;20(21):5345. doi: 10.3390/ijms20215345. Int J Mol Sci. 2019. PMID: 31661765 Free PMC article.
-
Mucopolysaccharidosis type VII (Sly syndrome) - What do we know?Mol Genet Metab. 2024 Mar;141(3):108145. doi: 10.1016/j.ymgme.2024.108145. Epub 2024 Jan 17. Mol Genet Metab. 2024. PMID: 38301529 Review.
-
Prenatal diagnosis of mucopolysaccharidosis type VII facilitating treatment in neonatal period.Prenat Diagn. 2023 Nov;43(12):1567-1569. doi: 10.1002/pd.6455. Epub 2023 Nov 14. Prenat Diagn. 2023. PMID: 37964423
-
Clinical course of sly syndrome (mucopolysaccharidosis type VII).J Med Genet. 2016 Jun;53(6):403-18. doi: 10.1136/jmedgenet-2015-103322. Epub 2016 Feb 23. J Med Genet. 2016. PMID: 26908836 Free PMC article.
-
The first mucopolysaccharidosis type VII in a Taiwanese girl: A case report and review of the literature.J Formos Med Assoc. 2022 Mar;121(3):712-717. doi: 10.1016/j.jfma.2021.07.024. Epub 2021 Aug 20. J Formos Med Assoc. 2022. PMID: 34420841 Review.
Cited by
-
Functions of Lysosomes in Mammalian Female Reproductive System.Reprod Dev Med. 2020 Apr;4(2):109-122. doi: 10.4103/2096-2924.288025. Reprod Dev Med. 2020. PMID: 40046839 Free PMC article.
-
Epidemiology of Mucopolysaccharidoses Update.Diagnostics (Basel). 2021 Feb 10;11(2):273. doi: 10.3390/diagnostics11020273. Diagnostics (Basel). 2021. PMID: 33578874 Free PMC article. Review.
-
Developmental odontogenic cysts with special focus on the occurrence of multiple cysts and syndromic association: a single-centre cross-sectional study from the Czech Republic.Orphanet J Rare Dis. 2025 Mar 4;20(1):103. doi: 10.1186/s13023-025-03623-5. Orphanet J Rare Dis. 2025. PMID: 40038706 Free PMC article.
-
Long-Term Follow-up Posthematopoietic Stem Cell Transplantation in a Japanese Patient with Type-VII Mucopolysaccharidosis.Diagnostics (Basel). 2020 Feb 16;10(2):105. doi: 10.3390/diagnostics10020105. Diagnostics (Basel). 2020. PMID: 32079065 Free PMC article.
-
First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation.Int J Mol Sci. 2019 Oct 28;20(21):5345. doi: 10.3390/ijms20215345. Int J Mol Sci. 2019. PMID: 31661765 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
