

Proteolysis of histidine kinase VgrS inhibits its autophosphorylation and promotes osmostress resistance in Xanthomonas campestris
- PMID: 30442885
- PMCID: PMC6237974
- DOI: 10.1038/s41467-018-07228-4
Proteolysis of histidine kinase VgrS inhibits its autophosphorylation and promotes osmostress resistance in Xanthomonas campestris
Abstract
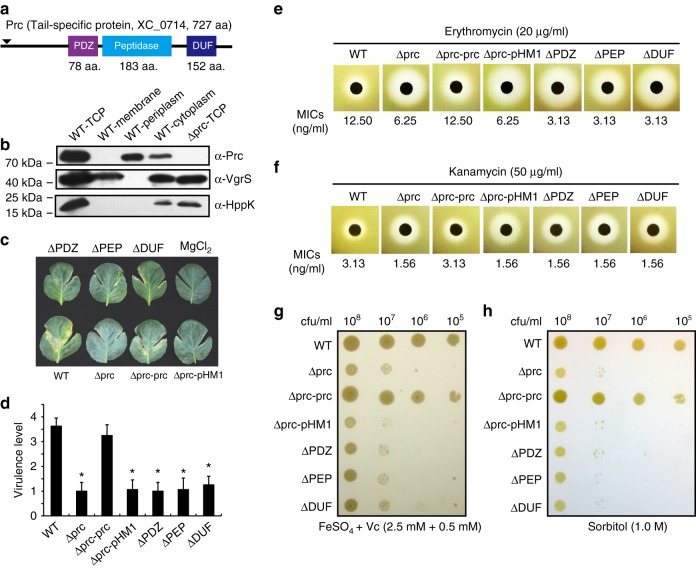
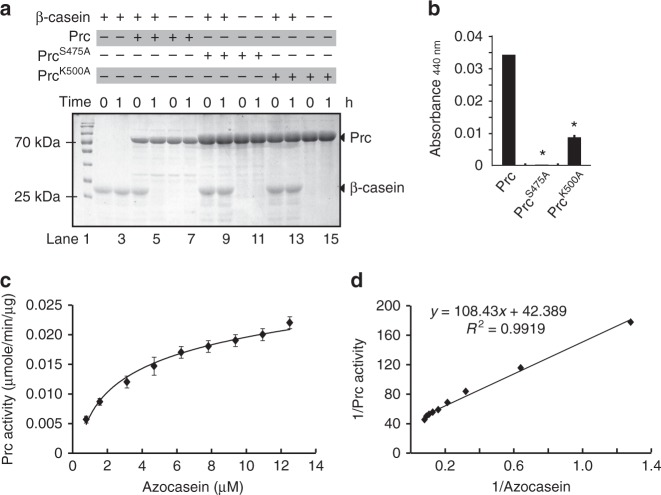
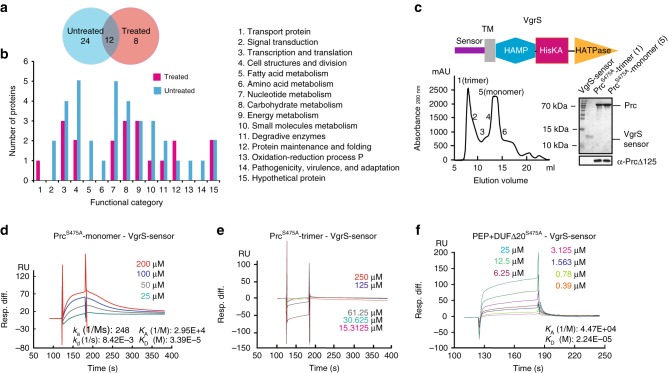
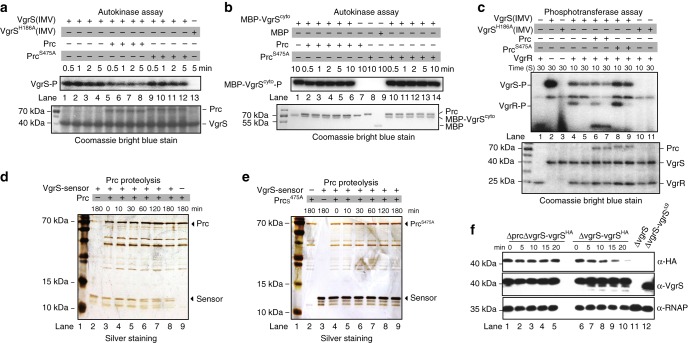
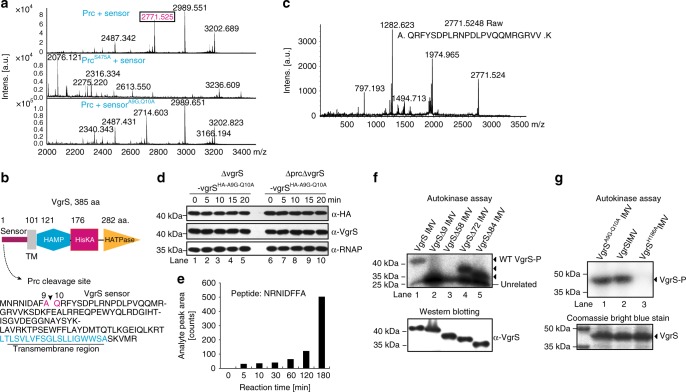
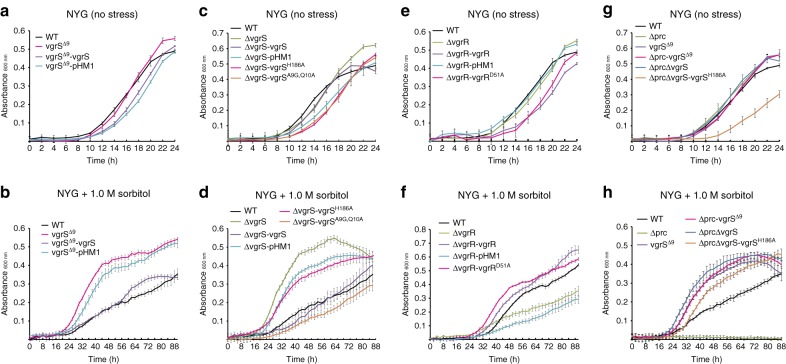
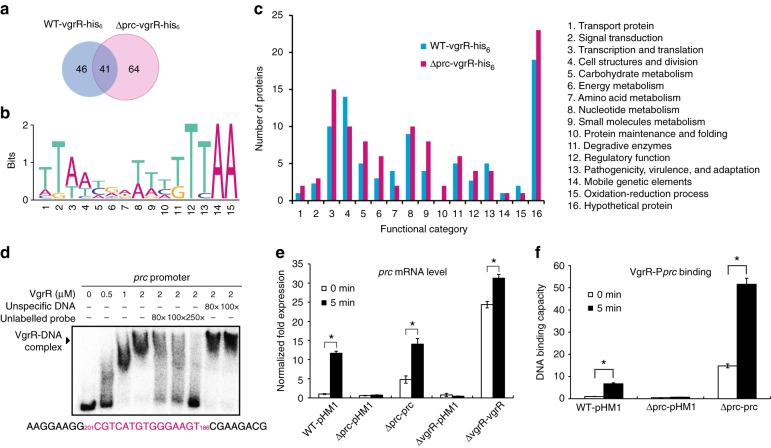
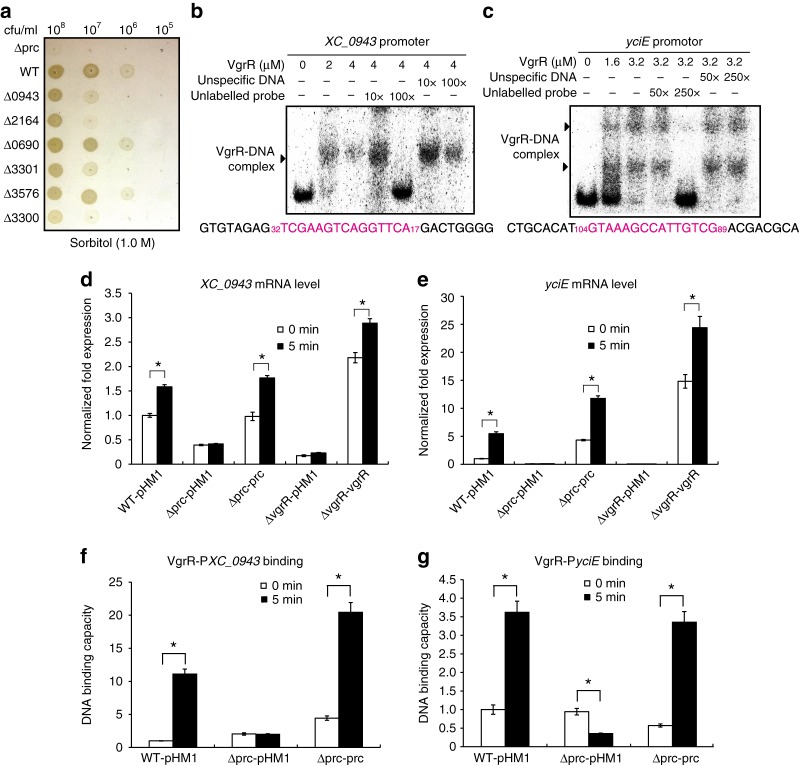
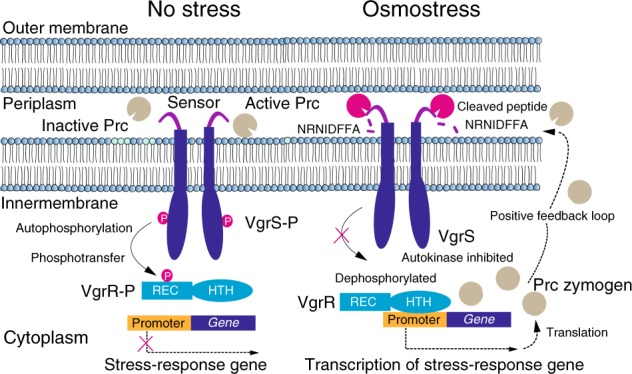
In bacterial cells, histidine kinases (HKs) are receptors that monitor environmental and intracellular stimuli. HKs and their cognate response regulators constitute two-component signalling systems (TCSs) that modulate cellular homeostasis through reversible protein phosphorylation. Here the authors show that the plant pathogen Xanthomonas campestris pv. campestris responds to osmostress conditions by regulating the activity of a HK (VgrS) via irreversible, proteolytic modification. This regulation is mediated by a periplasmic, PDZ-domain-containing protease (Prc) that cleaves the N-terminal sensor region of VgrS. Cleavage of VgrS inhibits its autokinase activity and regulates the ability of the cognate response regulator (VgrR) to bind promoters of downstream genes, thus promoting bacterial adaptation to osmostress.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Two-Component Signaling System VgrRS Directly Senses Extracytoplasmic and Intracellular Iron to Control Bacterial Adaptation under Iron Depleted Stress.PLoS Pathog. 2016 Dec 30;12(12):e1006133. doi: 10.1371/journal.ppat.1006133. eCollection 2016 Dec. PLoS Pathog. 2016. PMID: 28036380 Free PMC article.
-
The roles of histidine kinases in sensing host plant and cell-cell communication signal in a phytopathogenic bacterium.Philos Trans R Soc Lond B Biol Sci. 2019 Mar 4;374(1767):20180311. doi: 10.1098/rstb.2018.0311. Philos Trans R Soc Lond B Biol Sci. 2019. PMID: 30967026 Free PMC article. Review.
-
Fatty acid DSF binds and allosterically activates histidine kinase RpfC of phytopathogenic bacterium Xanthomonas campestris pv. campestris to regulate quorum-sensing and virulence.PLoS Pathog. 2017 Apr 3;13(4):e1006304. doi: 10.1371/journal.ppat.1006304. eCollection 2017 Apr. PLoS Pathog. 2017. PMID: 28369120 Free PMC article.
-
A three-component signalling system fine-tunes expression kinetics of HPPK responsible for folate synthesis by positive feedback loop during stress response of Xanthomonas campestris.Environ Microbiol. 2014 Jul;16(7):2126-44. doi: 10.1111/1462-2920.12293. Epub 2013 Oct 31. Environ Microbiol. 2014. PMID: 24119200
-
Biofilm formation and dispersal in Xanthomonas campestris.Microbes Infect. 2004 May;6(6):623-9. doi: 10.1016/j.micinf.2004.01.013. Microbes Infect. 2004. PMID: 15158198 Review.
Cited by
-
P. aeruginosa CtpA protease adopts a novel activation mechanism to initiate the proteolytic process.EMBO J. 2024 Apr;43(8):1634-1652. doi: 10.1038/s44318-024-00069-6. Epub 2024 Mar 11. EMBO J. 2024. PMID: 38467832 Free PMC article.
-
SstF, a novel sulforaphane-sensing transcription factor of Xanthomonas campestris, is required for sulforaphane tolerance and virulence.Mol Plant Pathol. 2023 May;24(5):452-465. doi: 10.1111/mpp.13314. Epub 2023 Feb 24. Mol Plant Pathol. 2023. PMID: 36829260 Free PMC article.
-
Insights into the structure and function of the histidine kinase ComP from Bacillus amyloliquefaciens based on molecular modeling.Biosci Rep. 2022 Oct 28;42(10):BSR20220352. doi: 10.1042/BSR20220352. Biosci Rep. 2022. PMID: 36052710 Free PMC article.
-
A review of approaches to control bacterial leaf blight in rice.World J Microbiol Biotechnol. 2022 May 17;38(7):113. doi: 10.1007/s11274-022-03298-1. World J Microbiol Biotechnol. 2022. PMID: 35578069 Review.
-
The Xanthomonas RaxH-RaxR Two-Component Regulatory System Is Orthologous to the Zinc-Responsive Pseudomonas ColS-ColR System.Microorganisms. 2021 Jul 7;9(7):1458. doi: 10.3390/microorganisms9071458. Microorganisms. 2021. PMID: 34361895 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
