

An assessment of true and false positive detection rates of stepwise epistatic model selection as a function of sample size and number of markers
- PMID: 30443009
- PMCID: PMC6462028
- DOI: 10.1038/s41437-018-0162-2
An assessment of true and false positive detection rates of stepwise epistatic model selection as a function of sample size and number of markers
Abstract
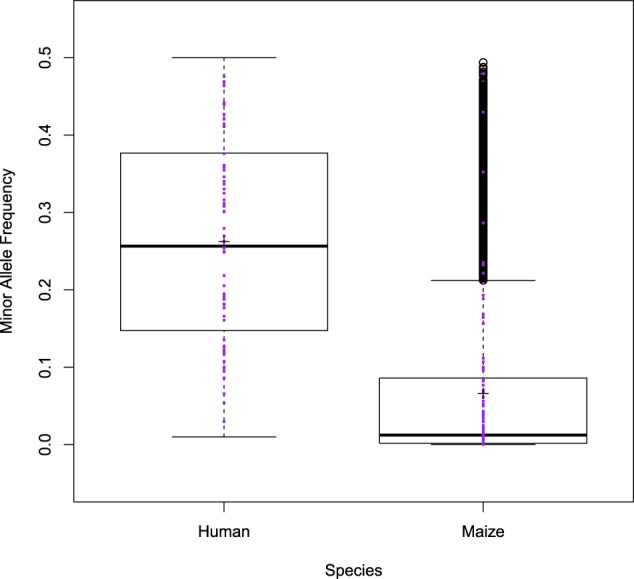
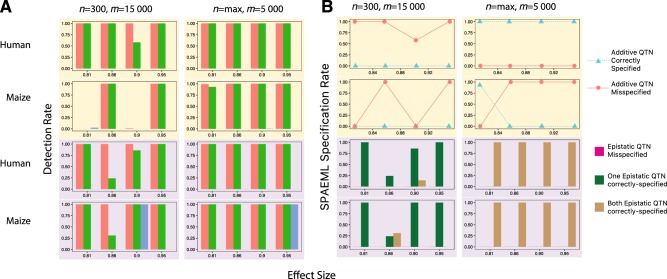
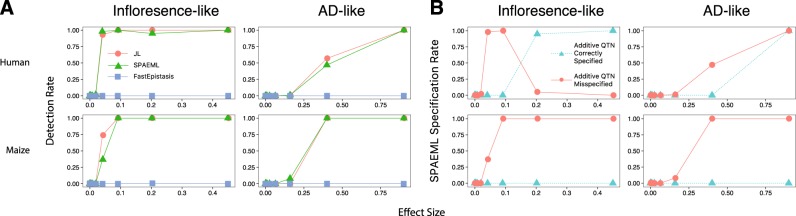
Association studies have been successful at identifying genomic regions associated with important traits, but routinely employ models that only consider the additive contribution of an individual marker. Because quantitative trait variability typically arises from multiple additive and non-additive sources, utilization of statistical approaches that include main and two-way interaction marker effects of several loci in one model could lead to unprecedented characterization of these sources. Here we examine the ability of one such approach, called the Stepwise Procedure for constructing an Additive and Epistatic Multi-Locus model (SPAEML), to detect additive and epistatic signals simulated using maize and human marker data. Our results revealed that SPAEML was capable of detecting quantitative trait nucleotides (QTNs) at sample sizes as low as n = 300 and consistently specifying signals as additive and epistatic for larger sizes. Sample size and minor allele frequency had a major influence on SPAEML's ability to distinguish between additive and epistatic signals, while the number of markers tested did not. We conclude that SPAEML is a useful approach for providing further elucidation of the additive and epistatic sources contributing to trait variability when applied to a small subset of genome-wide markers located within specific genomic regions identified using a priori analyses.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

Similar articles
-
A Genome-Wide Association Analysis Reveals Epistatic Cancellation of Additive Genetic Variance for Root Length in Arabidopsis thaliana.PLoS Genet. 2015 Sep 23;11(9):e1005541. doi: 10.1371/journal.pgen.1005541. eCollection 2015. PLoS Genet. 2015. PMID: 26397943 Free PMC article.
-
Factors influencing QTL mapping accuracy under complicated genetic models by computer simulation.Genet Mol Res. 2016 Dec 19;15(4). doi: 10.4238/gmr15049153. Genet Mol Res. 2016. PMID: 28002599
-
Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations.Theor Appl Genet. 2008 Jan;116(2):243-60. doi: 10.1007/s00122-007-0663-5. Epub 2007 Nov 6. Theor Appl Genet. 2008. PMID: 17985112
-
QTL-based evidence for the role of epistasis in evolution.Genet Res. 2005 Oct;86(2):89-95. doi: 10.1017/S0016672305007780. Genet Res. 2005. PMID: 16356282 Review.
-
Detecting epistatic interactions contributing to quantitative traits.Genet Epidemiol. 2004 Sep;27(2):141-52. doi: 10.1002/gepi.20006. Genet Epidemiol. 2004. PMID: 15305330 Review.
Cited by
-
Dissecting Adaptive Traits with Nested Association Mapping: Genetic Architecture of Inflorescence Morphology in Sorghum.G3 (Bethesda). 2020 May 4;10(5):1785-1796. doi: 10.1534/g3.119.400658. G3 (Bethesda). 2020. PMID: 32217633 Free PMC article.
-
Stimulation of distinct parietal locations differentiates frontal versus hippocampal network involvement in memory formation.Curr Res Neurobiol. 2022 Feb 17;3:100030. doi: 10.1016/j.crneur.2022.100030. eCollection 2022. Curr Res Neurobiol. 2022. PMID: 36518334 Free PMC article.
-
Epistasis Detection and Modeling for Genomic Selection in Cowpea (Vigna unguiculata L. Walp.).Front Genet. 2019 Jul 30;10:677. doi: 10.3389/fgene.2019.00677. eCollection 2019. Front Genet. 2019. PMID: 31417604 Free PMC article.
References
-
- Belcher AR, Cuesta-Marcos A, Smith KP, Mundt CC, Chen XM, Hayes PM. TCAP FAC-WIN6 elite barley GWAS panel QTL. I. barley stripe rust resistance QTL in facultative and winter six-rowed malt barley breeding programs identified via GWAS. Crop Sci. 2018;58(1):103–119. doi: 10.2135/cropsci2017.03.0206. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
