

Control and dysregulation of redox signalling in the gastrointestinal tract
- PMID: 30443019
- PMCID: PMC7919748
- DOI: 10.1038/s41575-018-0079-5
Control and dysregulation of redox signalling in the gastrointestinal tract
Abstract
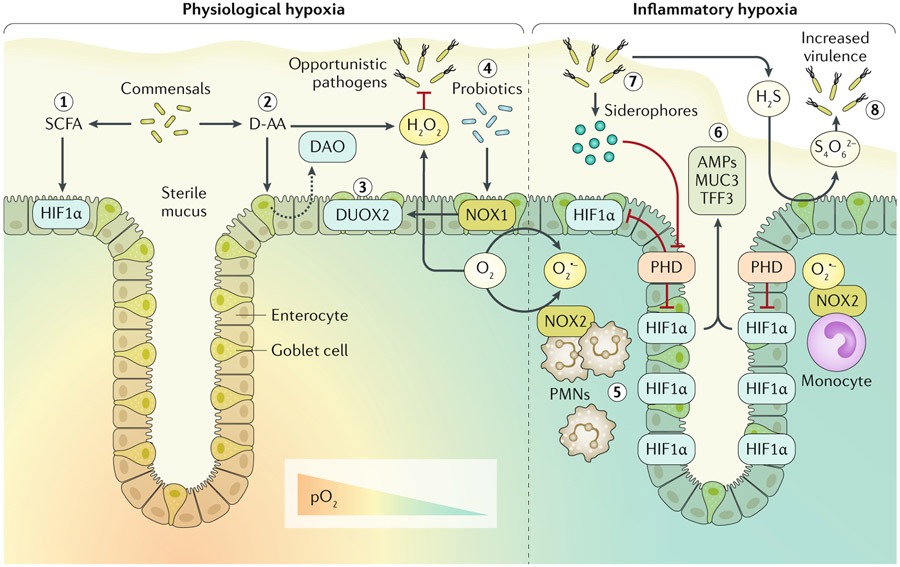
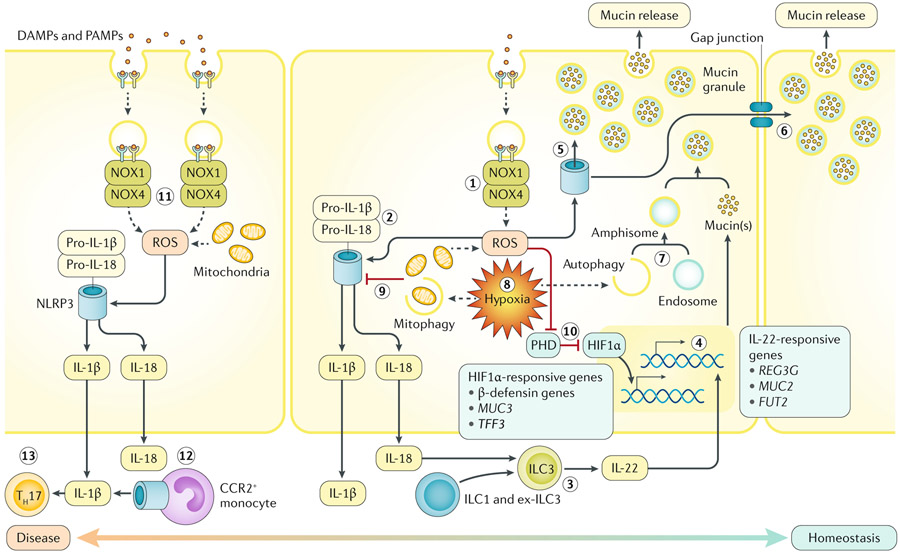
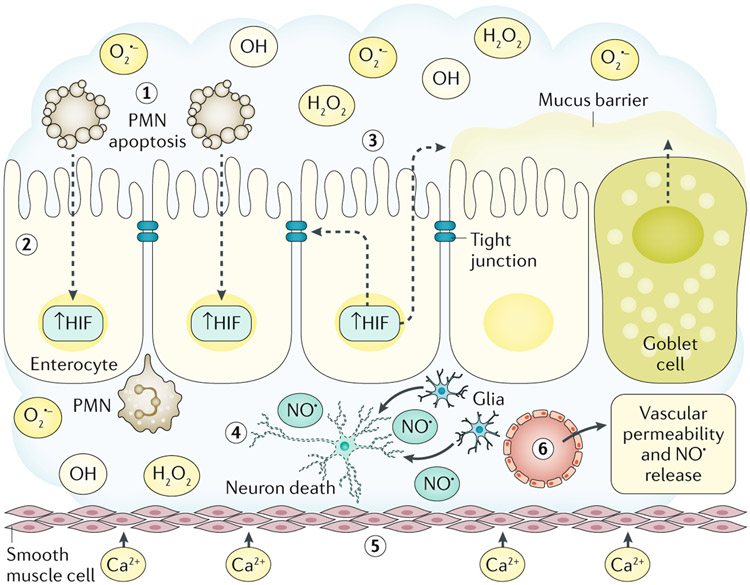
Redox signalling in the gastrointestinal mucosa is held in an intricate balance. Potent microbicidal mechanisms can be used by infiltrating immune cells, such as neutrophils, to protect compromised mucosae from microbial infection through the generation of reactive oxygen species. Unchecked, collateral damage to the surrounding tissue from neutrophil-derived reactive oxygen species can be detrimental; thus, maintenance and restitution of a breached intestinal mucosal barrier are paramount to host survival. Redox reactions and redox signalling have been studied for decades with a primary focus on contributions to disease processes. Within the past decade, an upsurge of exciting findings have implicated subtoxic levels of oxidative stress in processes such as maintenance of mucosal homeostasis, the control of protective inflammation and even regulation of tissue wound healing. Resident gut microbial communities have been shown to trigger redox signalling within the mucosa, which expresses similar but distinct enzymes to phagocytes. At the fulcrum of this delicate balance is the colonic mucosal epithelium, and emerging evidence suggests that precise control of redox signalling by these barrier-forming cells may dictate the outcome of an inflammatory event. This Review will address both the spectrum and intensity of redox activity pertaining to host-immune and host-microbiota crosstalk during homeostasis and disease processes in the gastrointestinal tract.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
