

Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome
- PMID: 30443250
- PMCID: PMC6223101
- DOI: 10.3389/fimmu.2018.02411
Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome
Abstract
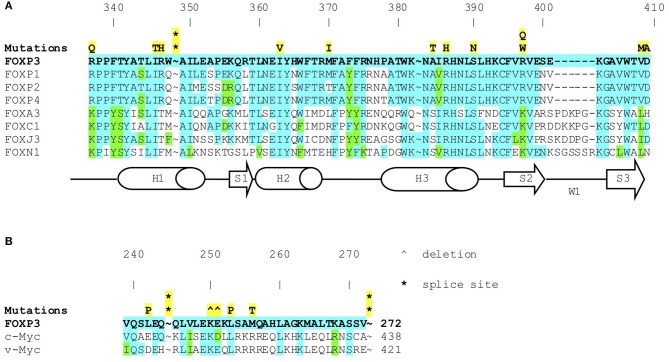
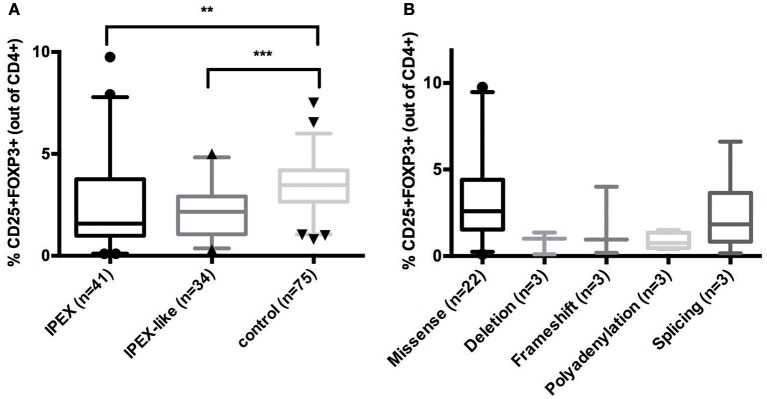
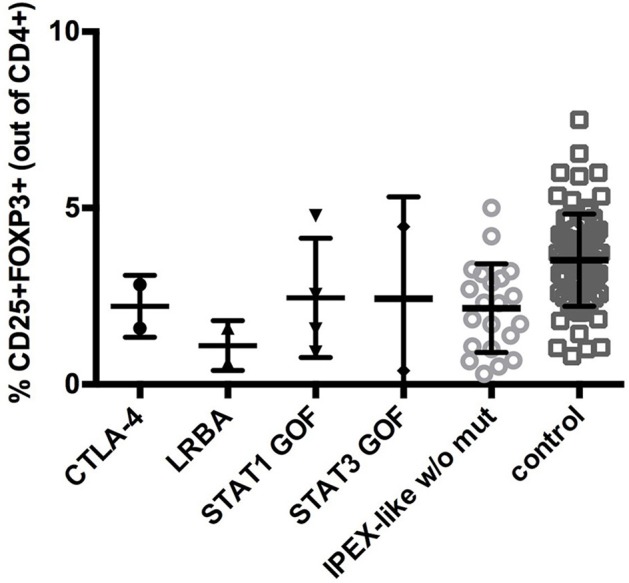
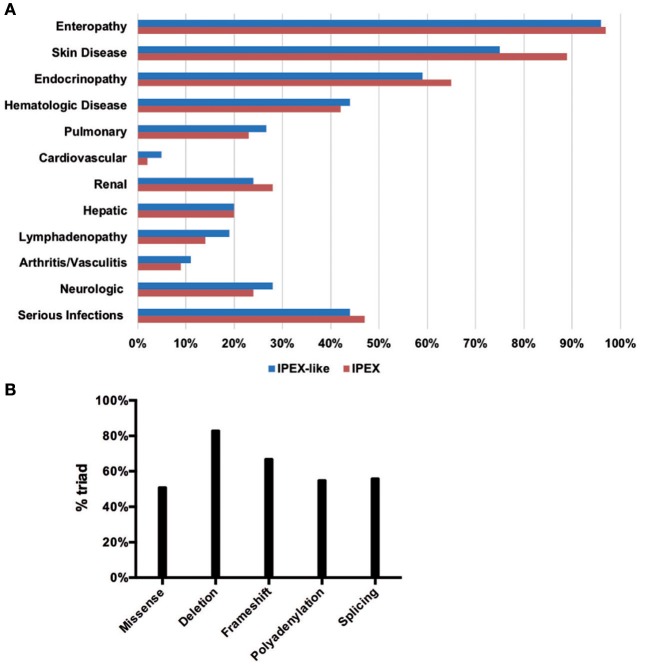
Background: Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-linked (IPEX) Syndrome is a rare recessive disorder caused by mutations in the FOXP3 gene. In addition, there has been an increasing number of patients with wild-type FOXP3 gene and, in some cases, mutations in other immune regulatory genes. Objective: To molecularly asses a cohort of 173 patients with the IPEX phenotype and to delineate the relationship between the clinical/immunologic phenotypes and the genotypes. Methods: We reviewed the clinical presentation and laboratory characteristics of each patient and compared clinical and laboratory data of FOXP3 mutation-positive (IPEX patients) with those from FOXP3 mutation-negative patients (IPEX-like). A total of 173 affected patients underwent direct sequence analysis of the FOXP3 gene while 85 IPEX-like patients with normal FOXP3 were investigated by a multiplex panel of "Primary Immune Deficiency (PID-related) genes." Results: Forty-four distinct FOXP3 variants were identified in 88 IPEX patients, 9 of which were not previously reported. Among the 85 IPEX-like patients, 19 different disease-associated variants affecting 9 distinct genes were identified. Conclusions: We provide a comprehensive analysis of the clinical features and molecular bases of IPEX and IPEX-like patients. Although we were not able to identify major distinctive clinical features to differentiate IPEX from IPEX-like syndromes, we propose a simple flow-chart to effectively evaluate such patients and to focus on the most likely molecular diagnosis. Given the large number of potential candidate genes and overlapping phenotypes, selecting a panel of PID-related genes will facilitate a molecular diagnosis.
Keywords: FOXP3; IPEX-like; X-linked (IPEX); enteropathy; immune dysregulation; immune regulatory genes; polyendocrinopathy; regulatory T cells.
Figures

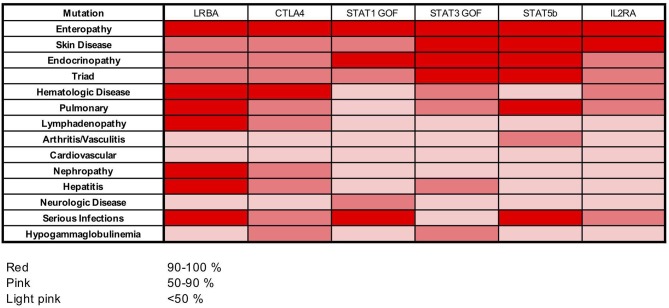
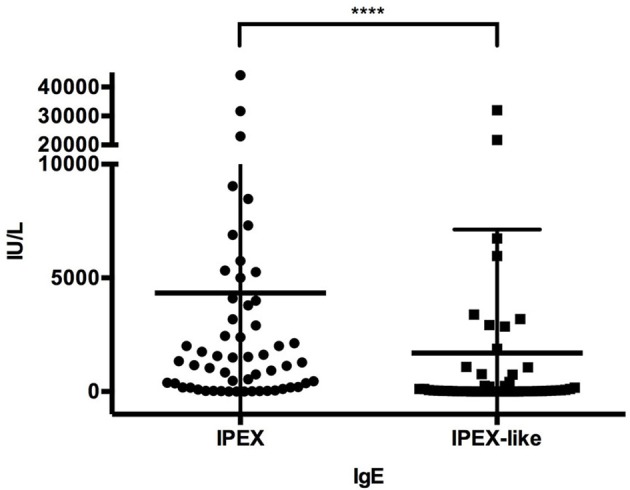
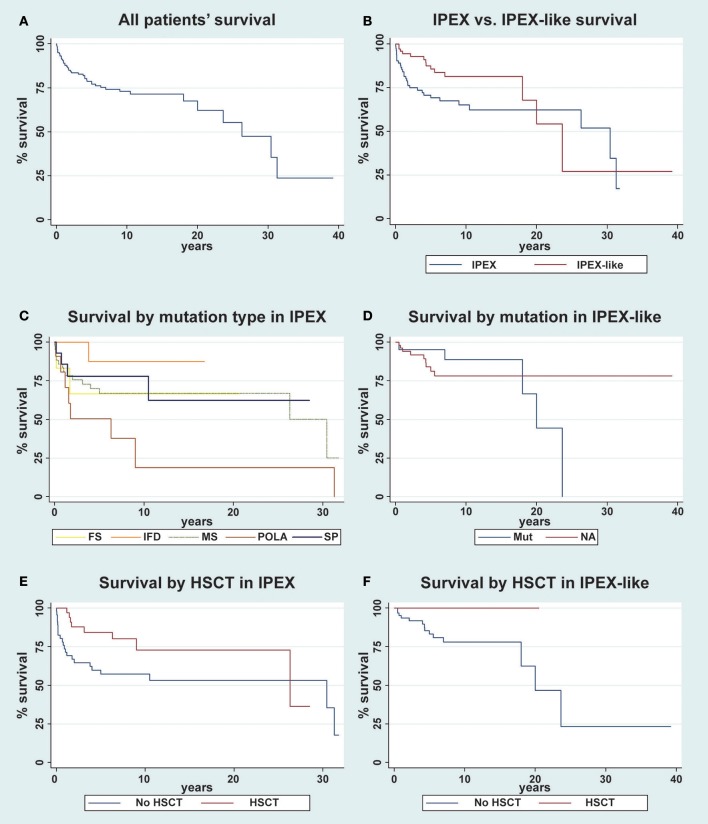
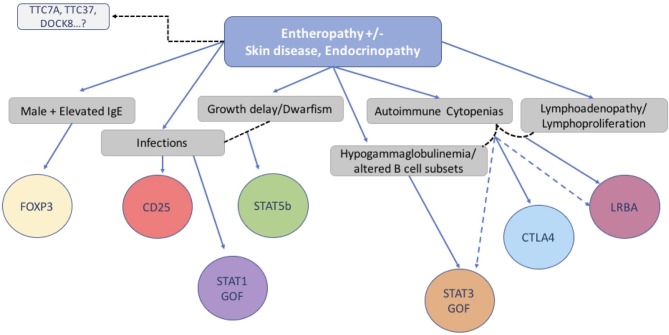
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical