

Evaluating the Information Content of Shallow Shotgun Metagenomics
- PMID: 30443602
- PMCID: PMC6234283
- DOI: 10.1128/mSystems.00069-18
Evaluating the Information Content of Shallow Shotgun Metagenomics
Abstract
Although microbial communities are associated with human, environmental, plant, and animal health, there exists no cost-effective method for precisely characterizing species and genes in such communities. While deep whole-metagenome shotgun (WMS) sequencing provides high taxonomic and functional resolution, it is often prohibitively expensive for large-scale studies. The prevailing alternative, 16S rRNA gene amplicon (16S) sequencing, often does not resolve taxonomy past the genus level and provides only moderately accurate predictions of the functional profile; thus, there is currently no widely accepted approach to affordable, high-resolution, taxonomic, and functional microbiome analysis. To address this technology gap, we evaluated the information content of shallow shotgun sequencing with as low as 0.5 million sequences per sample as an alternative to 16S sequencing for large human microbiome studies. We describe a library preparation protocol enabling shallow shotgun sequencing at approximately the same per-sample cost as 16S sequencing. We analyzed multiple real and simulated biological data sets, including two novel human stool samples with ultradeep sequencing of 2.5 billion sequences per sample, and found that shallow shotgun sequencing recovers more-accurate species-level taxonomic and functional profiles of the human microbiome than 16S sequencing. We discuss the inherent limitations of shallow shotgun sequencing and note that 16S sequencing remains a valuable and important method for taxonomic profiling of novel environments. Although deep WMS sequencing remains the gold standard for high-resolution microbiome analysis, we recommend that researchers consider shallow shotgun sequencing as a useful alternative to 16S sequencing for large-scale human microbiome research studies where WMS sequencing may be cost-prohibitive. IMPORTANCE A common refrain in recent microbiome-related academic meetings is that the field needs to move away from broad taxonomic surveys using 16S sequencing and toward more powerful longitudinal studies using shotgun sequencing. However, performing deep shotgun sequencing in large longitudinal studies remains prohibitively expensive for all but the most well-funded research labs and consortia, which leads many researchers to choose 16S sequencing for large studies, followed by deep shotgun sequencing on a subset of targeted samples. Here, we show that shallow- or moderate-depth shotgun sequencing may be used by researchers to obtain species-level taxonomic and functional data at approximately the same cost as amplicon sequencing. While shallow shotgun sequencing is not intended to replace deep shotgun sequencing for strain-level characterization, we recommend that microbiome scientists consider using shallow shotgun sequencing instead of 16S sequencing for large-scale human microbiome studies.
Keywords: human microbiome; metagenomics; microbiome; shotgun metagenomics.
Figures
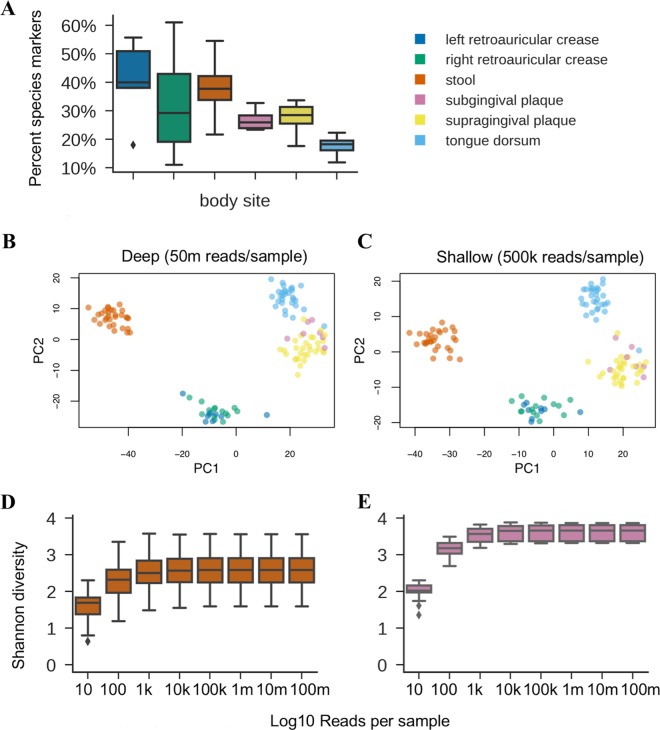
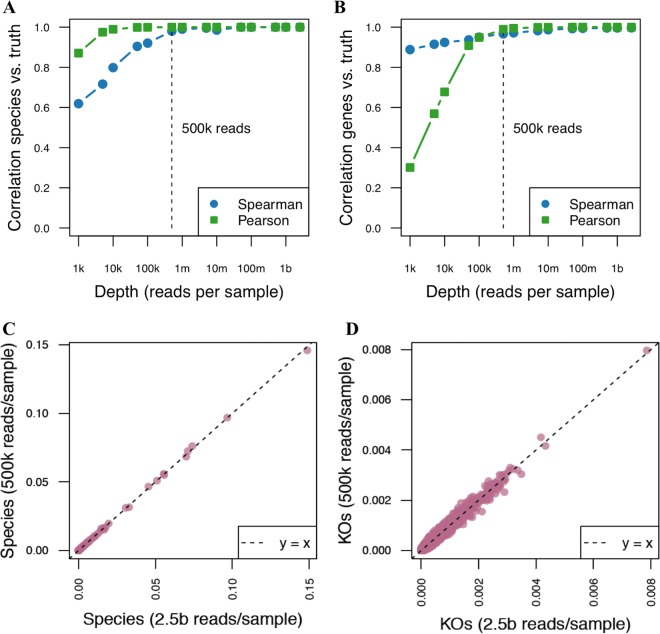
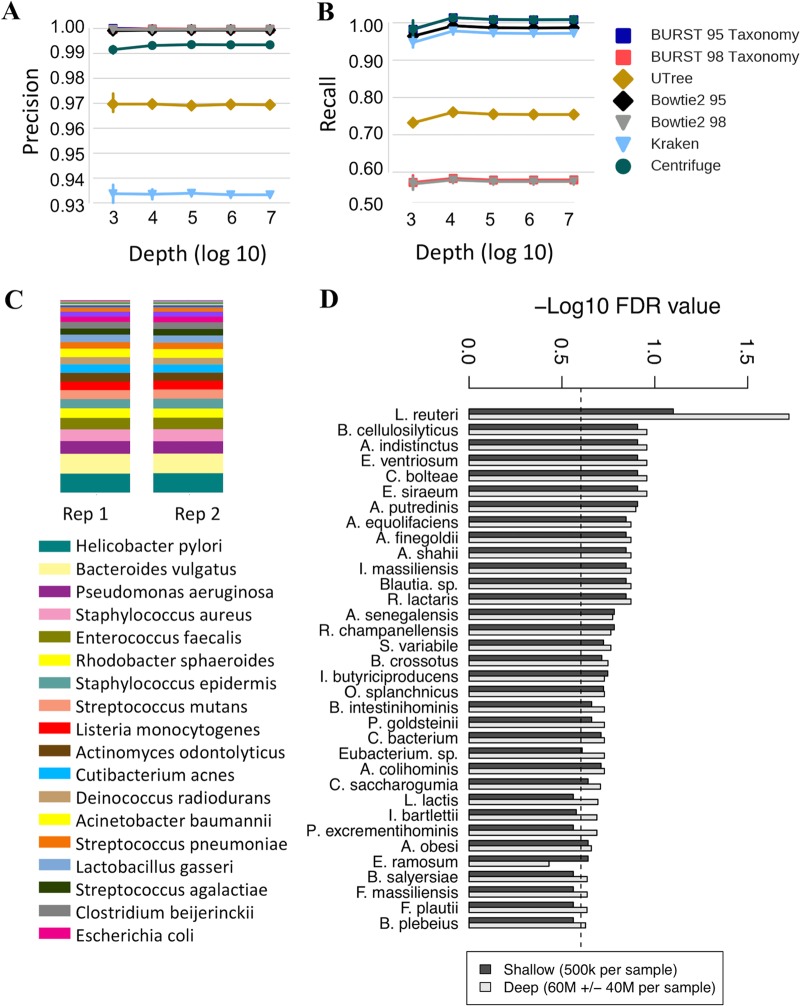
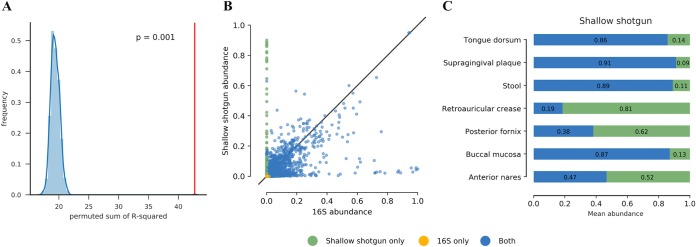
References
-
- Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vázquez-Baeza Y, González A, Morton JT, Mirarab S, Zech Xu Z, Jiang L, Haroon MF, Kanbar J, Zhu Q, Jin Song S, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, Earth Microbiome Project Consortium. 2017. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551:457–463. doi:10.1038/nature24621.] - DOI - PMC - PubMed
-
- Huttenhower C, Gevers D, Knight R, Abubucker S, Badger J, Chinwalla A, Creasy H, Earl A, FitzGerald M, Fulton R, Giglio M, Hallsworth-Pepin K, Lobos E, Madupu R, Magrini V, Martin J, Mitreva M, Muzny D, Sodergren E, Versalovic J, Wollam A, Worley K, Wortman J, Young S, Zeng Q, Aagaard K, Abolude O, Allen-Vercoe E, Alm E, Alvarado L, Andersen G, Anderson S, Appelbaum E, Arachchi H, Armitage G, Arze C, Ayvaz T, Baker C, Begg L, Belachew T, Bhonagiri V, Bihan M, Blaser M, Bloom T, Bonazzi V, Brooks J, Buck G, Buhay C, Busam D, Campbell J, et al. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi:10.1038/nature11234. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
