

Glutamatergic Neurotransmission: Pathway to Developing Novel Rapid-Acting Antidepressant Treatments
- PMID: 30445512
- PMCID: PMC6368372
- DOI: 10.1093/ijnp/pyy094
Glutamatergic Neurotransmission: Pathway to Developing Novel Rapid-Acting Antidepressant Treatments
Abstract
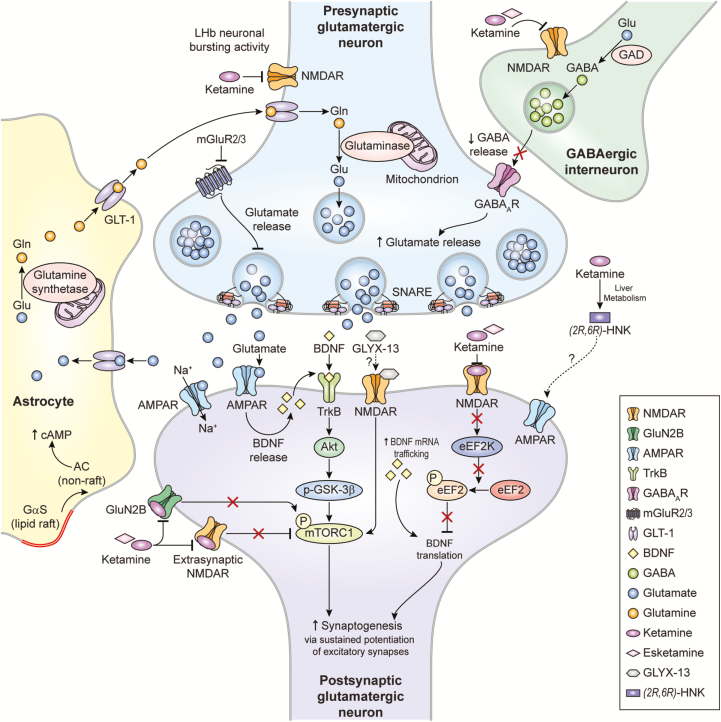
The underlying neurobiological basis of major depressive disorder remains elusive due to the severity, complexity, and heterogeneity of the disorder. While the traditional monoaminergic hypothesis has largely fallen short in its ability to provide a complete picture of major depressive disorder, emerging preclinical and clinical findings suggest that dysfunctional glutamatergic neurotransmission may underlie the pathophysiology of both major depressive disorder and bipolar depression. In particular, recent studies showing that a single intravenous infusion of the glutamatergic modulator ketamine elicits fast-acting, robust, and relatively sustained antidepressant, antisuicidal, and antianhedonic effects in individuals with treatment-resistant depression have prompted tremendous interest in understanding the mechanisms responsible for ketamine's clinical efficacy. These results, coupled with new evidence of the mechanistic processes underlying ketamine's effects, have led to inventive ways of investigating, repurposing, and expanding research into novel glutamate-based therapeutic targets with superior antidepressant effects but devoid of dissociative side effects. Ketamine's targets include noncompetitive N-methyl-D-aspartate receptor inhibition, α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid throughput potentiation coupled with downstream signaling changes, and N-methyl-D-aspartate receptor targets localized on gamma-aminobutyric acid-ergic interneurons. Here, we review ketamine and other potentially novel glutamate-based treatments for treatment-resistant depression, including N-methyl-D-aspartate receptor antagonists, glycine binding site ligands, metabotropic glutamate receptor modulators, and other glutamatergic modulators. Both the putative mechanisms of action of these agents and clinically relevant studies are described.
Figures
Similar articles
-
A review of ketamine in affective disorders: current evidence of clinical efficacy, limitations of use and pre-clinical evidence on proposed mechanisms of action.J Affect Disord. 2014 Mar;156:24-35. doi: 10.1016/j.jad.2013.11.014. Epub 2013 Dec 10. J Affect Disord. 2014. PMID: 24388038 Review.
-
Acute subanesthetic ketamine-induced effects on the mismatch negativity and their relationship to early and sustained treatment response in major depressive disorder.J Psychopharmacol. 2025 Jun;39(6):577-592. doi: 10.1177/02698811251319456. Epub 2025 Feb 26. J Psychopharmacol. 2025. PMID: 40012166 Free PMC article. Clinical Trial.
-
Ketamine for Treatment-Resistant Depression: a New Advocate.Rev Invest Clin. 2018;70(2):65-7. doi: 10.24875/RIC.18002501. Rev Invest Clin. 2018. PMID: 29718013
-
Ketamine: The final frontier or another depressing end?Behav Brain Res. 2020 Apr 6;383:112508. doi: 10.1016/j.bbr.2020.112508. Epub 2020 Feb 1. Behav Brain Res. 2020. PMID: 32017978 Free PMC article. Review.
-
Overlap in the neural circuitry and molecular mechanisms underlying ketamine abuse and its use as an antidepressant.Behav Brain Res. 2020 Apr 20;384:112548. doi: 10.1016/j.bbr.2020.112548. Epub 2020 Feb 13. Behav Brain Res. 2020. PMID: 32061748 Free PMC article. Review.
Cited by
-
Dissecting the molecular mechanisms underlying the antidepressant activities of herbal medicines through the comprehensive review of the recent literatures.Front Psychiatry. 2022 Dec 22;13:1054726. doi: 10.3389/fpsyt.2022.1054726. eCollection 2022. Front Psychiatry. 2022. PMID: 36620687 Free PMC article. Review.
-
The stressed synapse 2.0: pathophysiological mechanisms in stress-related neuropsychiatric disorders.Nat Rev Neurosci. 2022 Feb;23(2):86-103. doi: 10.1038/s41583-021-00540-x. Epub 2021 Dec 10. Nat Rev Neurosci. 2022. PMID: 34893785 Review.
-
Ghrelin, Neuroinflammation, Oxidative Stress, and Mood Disorders: What Are the Connections?Curr Neuropharmacol. 2025;23(2):172-186. doi: 10.2174/1570159X22999240722095039. Curr Neuropharmacol. 2025. PMID: 39041263 Free PMC article. Review.
-
Novel Glutamatergic Modulators for the Treatment of Mood Disorders: Current Status.CNS Drugs. 2021 May;35(5):527-543. doi: 10.1007/s40263-021-00816-x. Epub 2021 Apr 26. CNS Drugs. 2021. PMID: 33904154 Free PMC article. Review.
-
A wake-up call: Sleep physiology and related translational discrepancies in studies of rapid-acting antidepressants.Prog Neurobiol. 2021 Nov;206:102140. doi: 10.1016/j.pneurobio.2021.102140. Epub 2021 Aug 14. Prog Neurobiol. 2021. PMID: 34403718 Free PMC article.
References
-
- Aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, Mathew SJ(2010)Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry 67:139–145. - PubMed
