

PXE, a Mysterious Inborn Error Clarified
- PMID: 30446375
- PMCID: PMC6340748
- DOI: 10.1016/j.tibs.2018.10.005
PXE, a Mysterious Inborn Error Clarified
Abstract
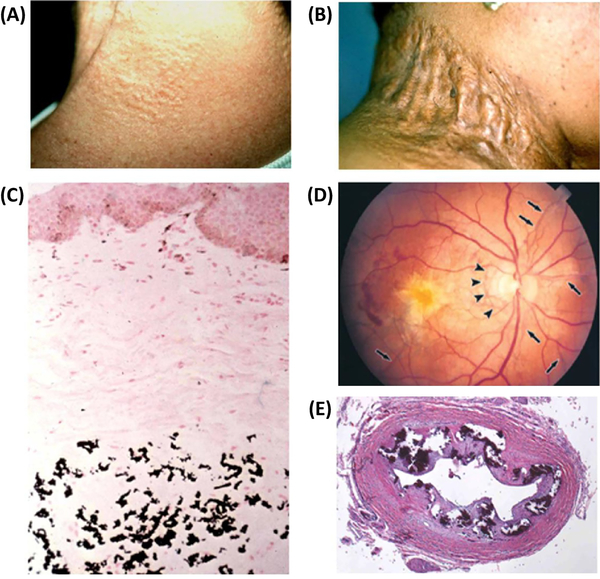
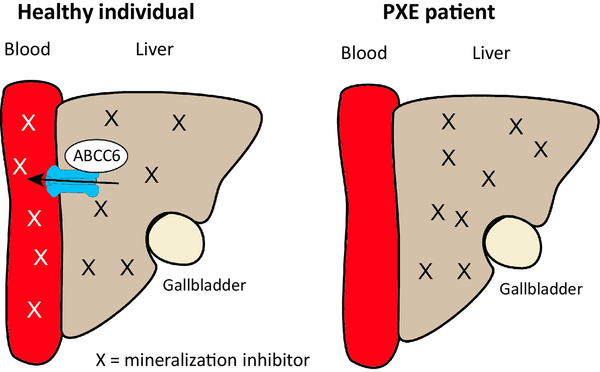
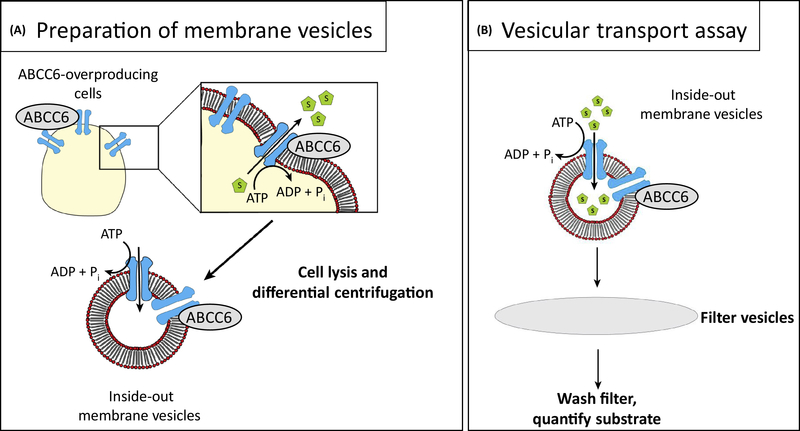
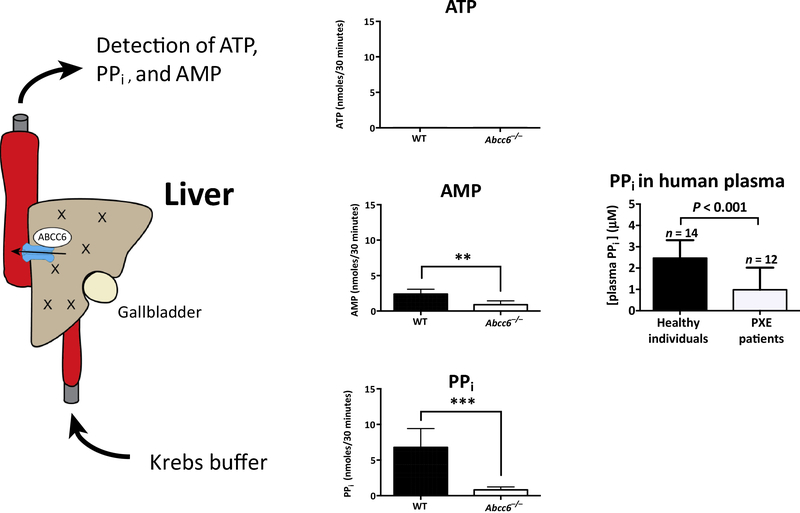
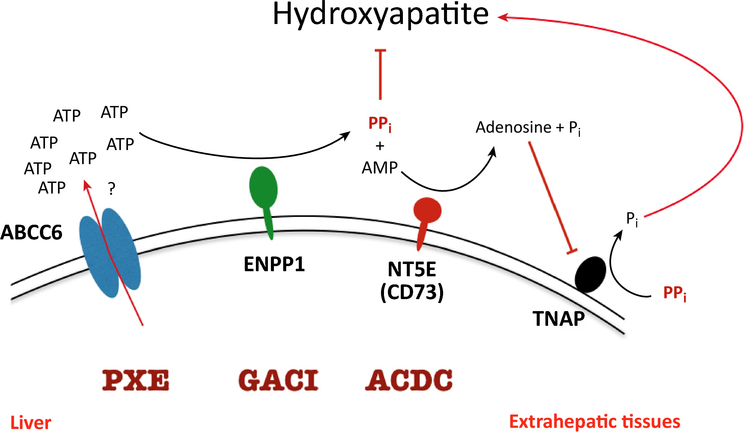
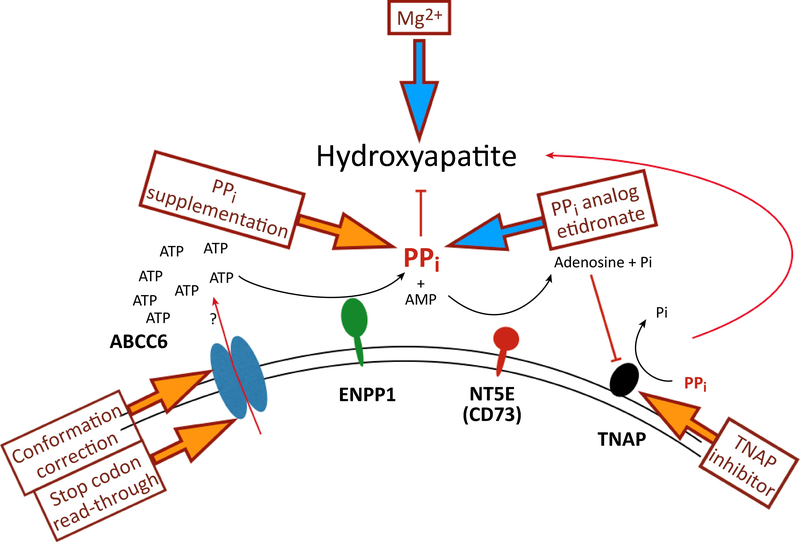
Ever since Garrod deduced the existence of inborn errors in 1901, a vast array of metabolic diseases has been identified and characterized in molecular terms. In 2018 it is difficult to imagine that there is any uncharted backyard left in the metabolic disease landscape. Nevertheless, it took until 2013 to identify the cause of a relatively frequent inborn error, pseudoxanthoma elasticum (PXE), a disorder resulting in aberrant calcification. The mechanism found was not only biochemically interesting but also points to possible new treatments for PXE, a disease that has remained untreatable. In this review we sketch the tortuous road that led to the biochemical understanding of PXE and to new ideas for treatment. We also discuss some of the controversies still haunting the field.
Keywords: ABC-transporters; ABCC6; Calcification; Pseudoxanthoma; Pyrophosphate.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Figures
References
-
- Le Boulanger G et al. (2009) An unusual severe vascular case of pseudoxanthoma elasticum presenting as generalized arterial calcification of infancy. Am. J. Med. Genet 152, 118–123 - PubMed
-
- Kranenburg G et al. (2018) Etidronate for Prevention of Ectopic Mineralization in Patients With Pseudoxanthoma Elasticum. J. Am. Coll. Cardiol 71, 1117–1126 - PubMed
-
- Le Saux O et al. (2000) Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet 25, 223–227 - PubMed
-
- Bergen AA et al. (2000) Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet 25, 228–231 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
