

Nicotine Modulates Growth Factors and MicroRNA to Promote Inflammatory and Fibrotic Processes
- PMID: 30446578
- PMCID: PMC6323623
- DOI: 10.1124/jpet.118.252650
Nicotine Modulates Growth Factors and MicroRNA to Promote Inflammatory and Fibrotic Processes
Abstract
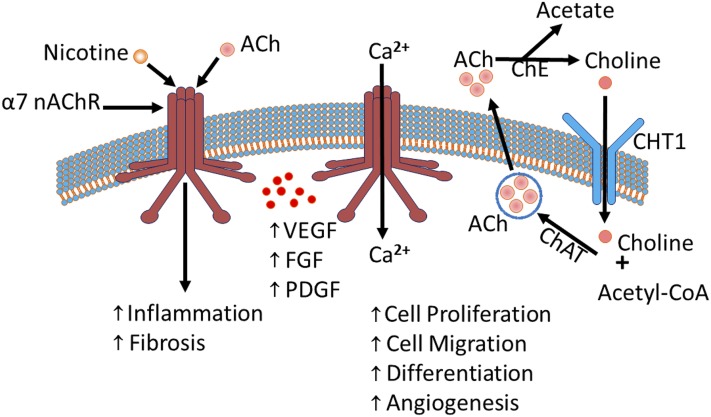
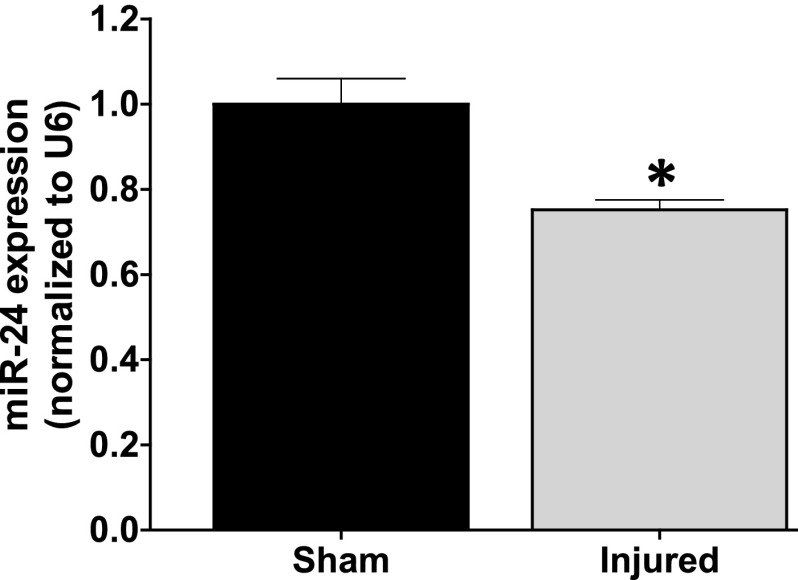
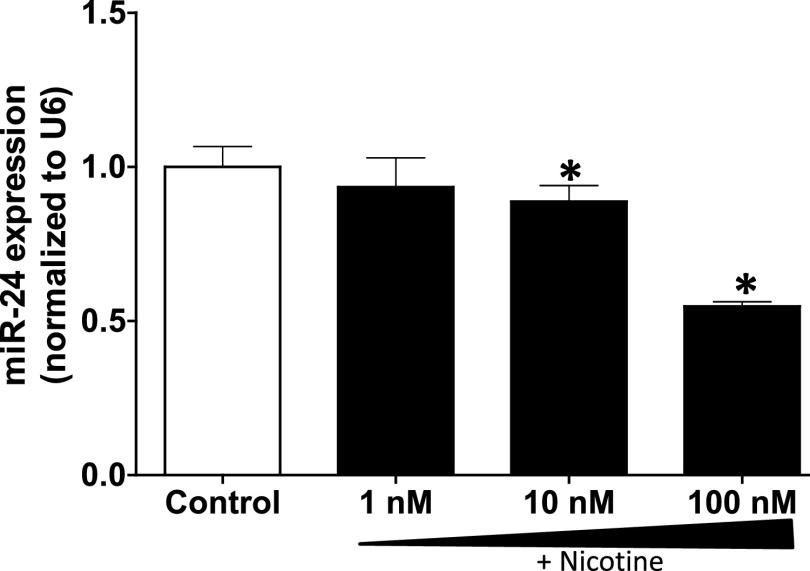
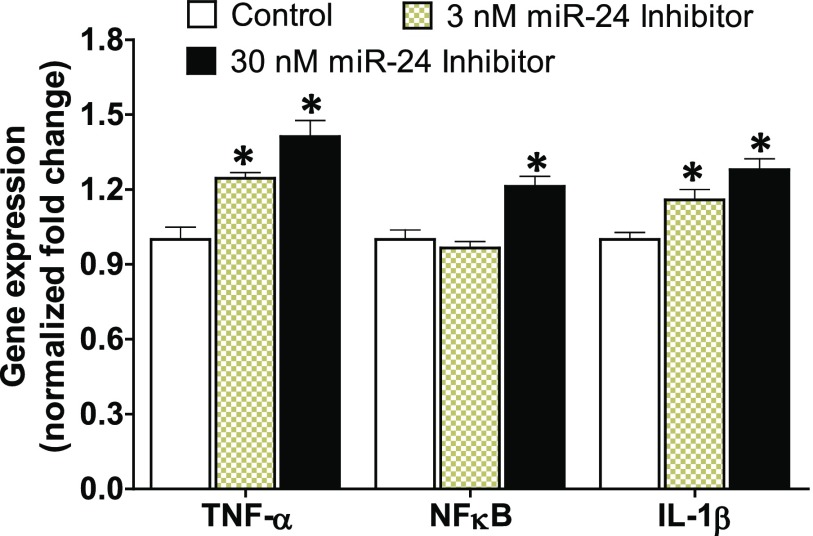
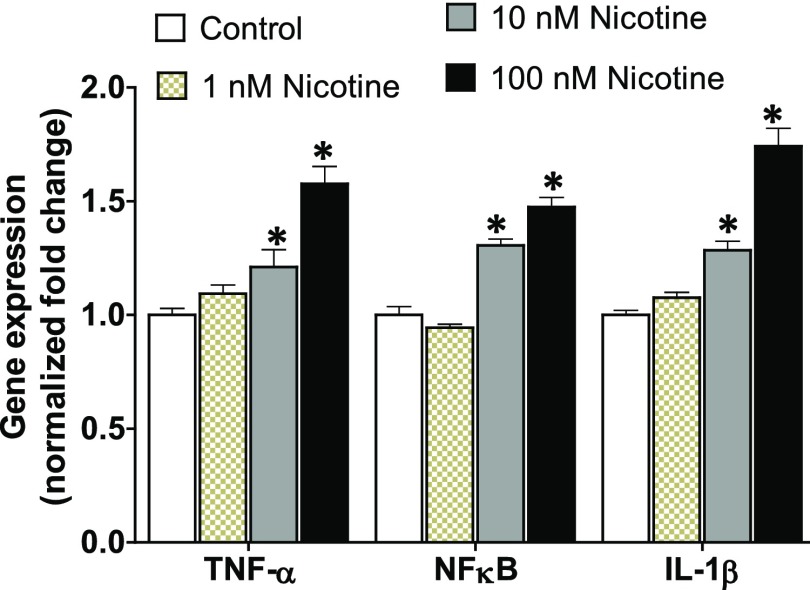
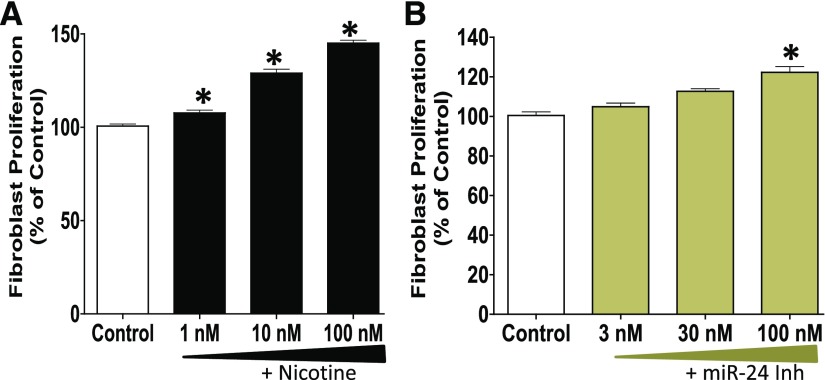
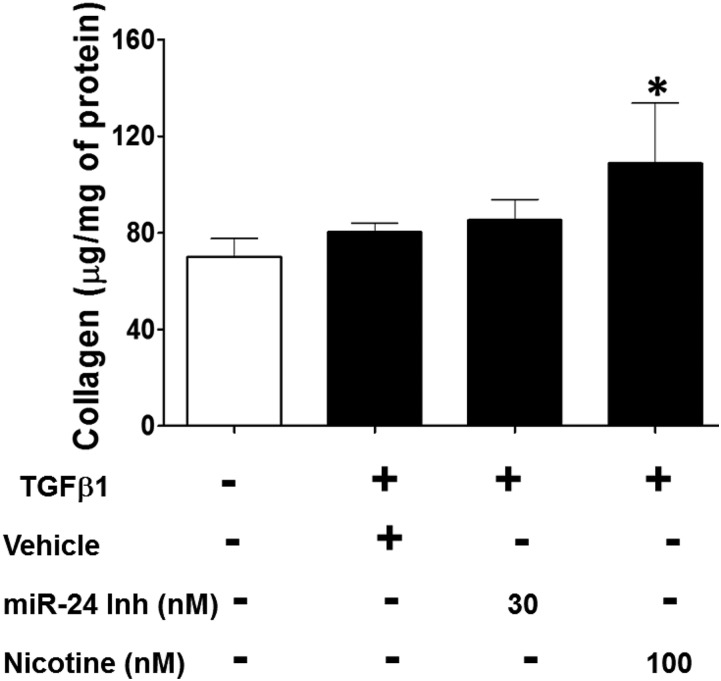
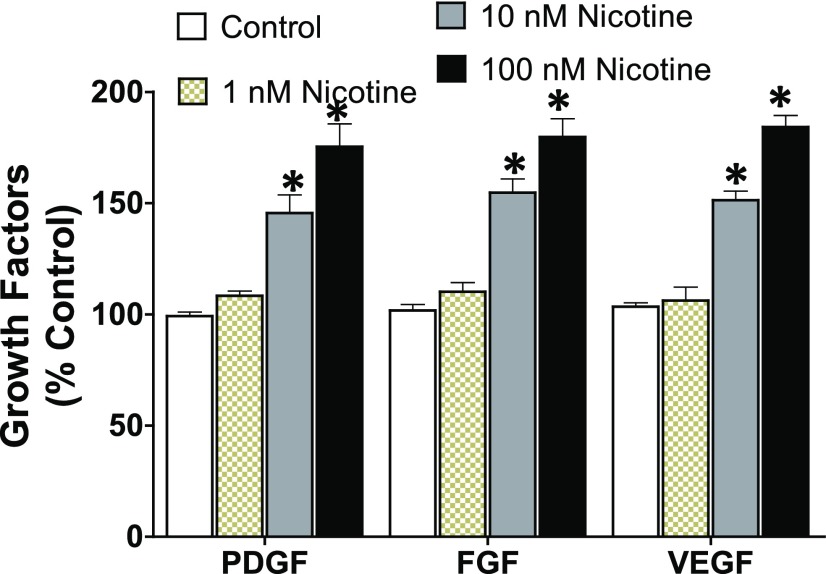
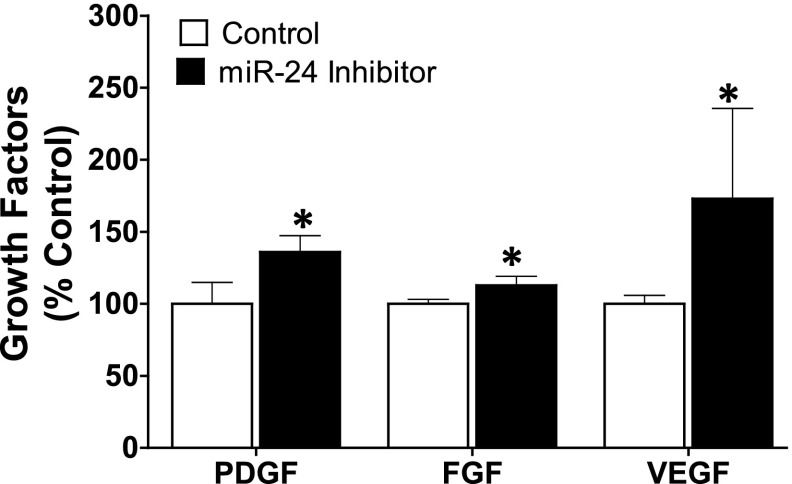
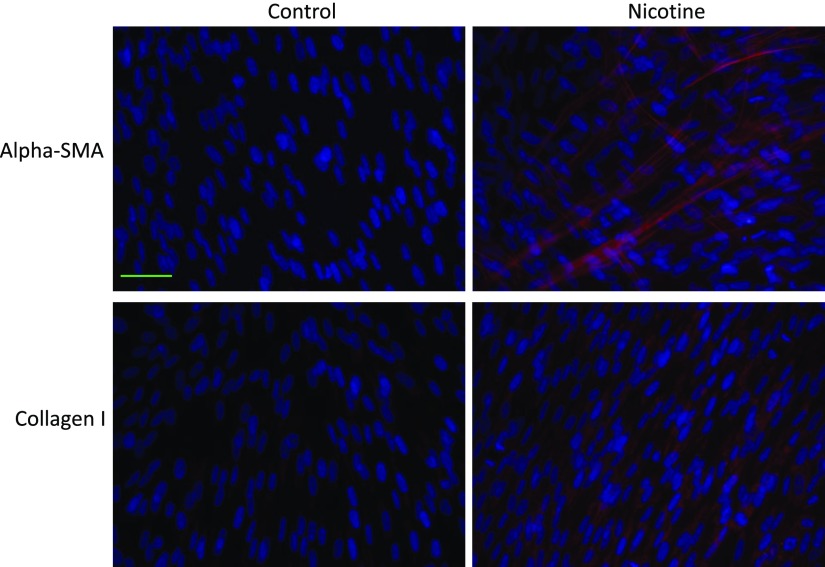
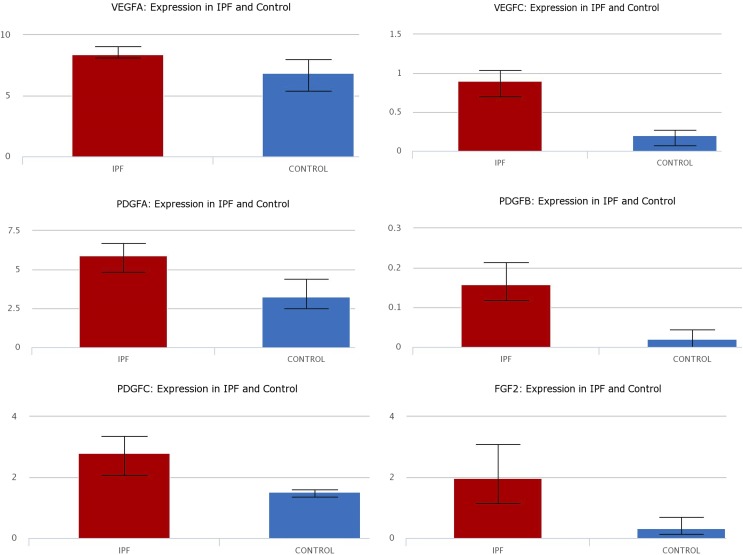
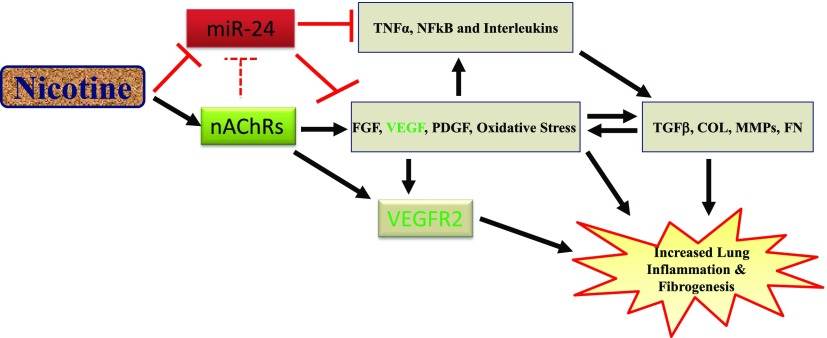
Idiopathic pulmonary fibrosis (IPF) is a fatal disease that destroys the structure and function of the lungs. Risk factors include advanced age and genetic predisposition. However, tobacco use is the chief modifiable risk factor. The prevalence of tobacco use in IPF reaches up to 80%. Although tobacco smoke contains over 5000 chemicals, nicotine is a major component. Nicotine is a bioactive molecule that acts upon nicotinic acetylcholine receptors expressed on neuronal and non-neuronal cells including endothelial cells. Accordingly, it has a pleiotropic effect on cell proliferation and angiogenesis. The angiogenic effect is partly mediated by stimulation of growth factors including fibroblast, platelet-derived, and vascular endothelial growth factors. Nintedanib, a Food and Drug Administration-approved drug for IPF, works by inhibiting receptors for these growth factors, suggesting a pathobiologic role of the growth factors in IPF and a potential mechanism by which tobacco use may exacerbate the disease process; additionally, nicotine downregulates anti-inflammatory microRNAs (miRs) in lung cells. Here, we profiled the expression of miRs in lung tissues explanted from a lung injury model and examined the effect of nicotine on one of the identified miRs (miR-24) and its downstream targets. Our data show that miR-24 is downregulated during lung injury and is suppressed by nicotine. We also found that nicotine upregulates the expression of inflammatory cytokines targeted by miR-24. Finally, nicotine stimulated growth factors, fibroblast proliferation, collagen release, and expression of myofibroblast markers. Taken together, nicotine, alone or as a component of tobacco smoke, may accelerate the disease process in IPF through stimulation of growth factors and downregulation of anti-inflammatory miRs.
Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
References
-
- Atamas SP. (2017) Vascular endothelial growth factor in idiopathic pulmonary fibrosis. An imbalancing act. Am J Respir Crit Care Med 196:409–411. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
