

Mutations in STAG2 cause an X-linked cohesinopathy associated with undergrowth, developmental delay, and dysmorphia: Expanding the phenotype in males
- PMID: 30447054
- PMCID: PMC6393687
- DOI: 10.1002/mgg3.501
Mutations in STAG2 cause an X-linked cohesinopathy associated with undergrowth, developmental delay, and dysmorphia: Expanding the phenotype in males
Abstract
Background: The cohesin complex is a multi-subunit protein complex which regulates sister chromatid cohesion and separation during cellular division. In addition, this evolutionarily conserved protein complex plays an integral role in DNA replication, DNA repair, and the regulation of transcription. The core complex is composed of four subunits: RAD21, SMC1A, SMC3, and STAG1/2. Mutations in these proteins have been implicated in human developmental disorders collectively termed "cohesinopathies."
Methods: Using clinical exome sequencing, we have previously identified three female cases with heterozygous STAG2 mutations and overlapping syndromic phenotypes. Subsequently, a familial missense variant was identified in five male family members.
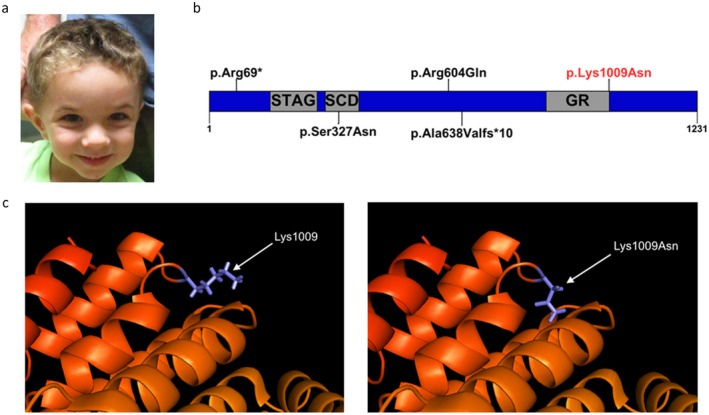
Results: We now present the case of a 4-year-old male with developmental delay, failure to thrive, short stature, and polydactyly with a likely pathogenic STAG2 de novo missense hemizygous variant, c.3027A>T, p.Lys1009Asn. Furthermore, we compare the phenotypes of the four previously reported STAG2 variants with our case.
Conclusion: We conclude that mutations in STAG2 cause a novel constellation of sex-specific cohesinopathy-related phenotypes and are furthermore, essential for neurodevelopment, human growth, and behavioral development.
Keywords: STAG2; X-linked gene; clinical exome sequencing; cohesin complex; cohesin-associated genes; cohesinopathies; human growth; neurodevelopment; reanalysis.
© 2018 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals, Inc.
Conflict of interest statement
No conflict of interests.
Figures
References
-
- Bonnet, C. , Leheup, B. , Beri, M. , Philippe, C. , Gregoire, M. J. , & Jonveaux, P. (2009). Aberrant GRIA3 transcripts with multi‐exon duplications in a family with X‐linked mental retardation. American Journal of Medical Genetics. Part A, 149A(6), 1280–1289. - PubMed
-
- Di Benedetto, D. , Di Vita, G. , Romano, C. , Giudice, M. L. , Vitello, G. A. , Zingale, M. , … Fichera, M. (2013). 6p22.3 deletion: Report of a patient with autism, severe intellectual disability and electroencephalographic anomalies. Molecular Cytogenetics, 6(1), 4 10.1186/1755-8166-6-4 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
