

Nonadiabatic dynamics: The SHARC approach
- PMID: 30450129
- PMCID: PMC6220962
- DOI: 10.1002/wcms.1370
Nonadiabatic dynamics: The SHARC approach
Abstract
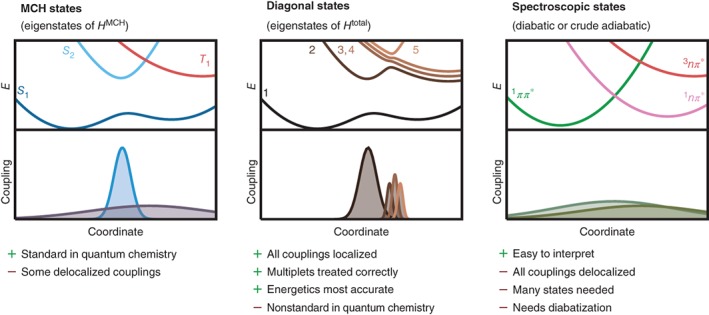
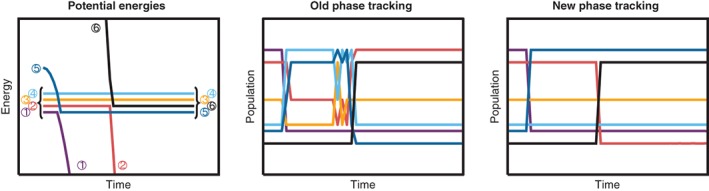
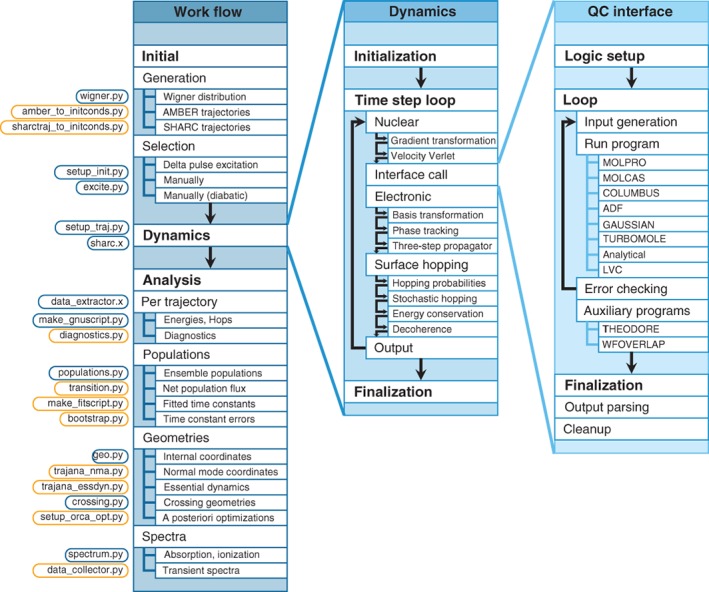
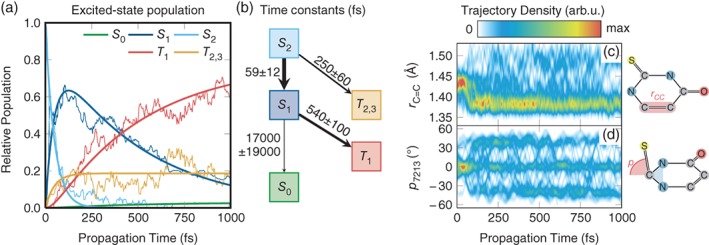
We review the Surface Hopping including ARbitrary Couplings (SHARC) approach for excited-state nonadiabatic dynamics simulations. As a generalization of the popular surface hopping method, SHARC allows simulating the full-dimensional dynamics of molecules including any type of coupling terms beyond nonadiabatic couplings. Examples of these arbitrary couplings include spin-orbit couplings or dipole moment-laser field couplings, such that SHARC can describe ultrafast internal conversion, intersystem crossing, and radiative processes. The key step of the SHARC approach consists of a diagonalization of the Hamiltonian including these couplings, such that the nuclear dynamics is carried out on potential energy surfaces including the effects of the couplings-this is critical in any applications considering, for example, transition metal complexes or strong laser fields. We also give an overview over the new SHARC2.0 dynamics software package, released under the GNU General Public License, which implements the SHARC approach and several analysis tools. The review closes with a brief survey of applications where SHARC was employed to study the nonadiabatic dynamics of a wide range of molecular systems. This article is categorized under: Theoretical and Physical Chemistry > Reaction Dynamics and KineticsSoftware > Simulation MethodsSoftware > Quantum Chemistry.
Keywords: Ab initio molecular dynamics; SHARC; excited states; nonadiabatic dynamics; surface hopping.
Figures
Similar articles
-
Surface hopping dynamics including intersystem crossing using the algebraic diagrammatic construction method.J Chem Phys. 2017 Nov 14;147(18):184109. doi: 10.1063/1.4999687. J Chem Phys. 2017. PMID: 29141436
-
SHARC: ab Initio Molecular Dynamics with Surface Hopping in the Adiabatic Representation Including Arbitrary Couplings.J Chem Theory Comput. 2011 May 10;7(5):1253-8. doi: 10.1021/ct1007394. Epub 2011 Mar 29. J Chem Theory Comput. 2011. PMID: 26610121
-
Internal conversion and intersystem crossing dynamics based on coupled potential energy surfaces with full geometry-dependent spin-orbit and derivative couplings. Nonadiabatic photodissociation dynamics of NH3(A) leading to the NH(X3Σ-, a1Δ) + H2 channel.Phys Chem Chem Phys. 2022 Jun 22;24(24):15060-15067. doi: 10.1039/d2cp01271e. Phys Chem Chem Phys. 2022. PMID: 35696936
-
Nonadiabatic Dynamics Simulations for Photoinduced Processes in Molecules and Semiconductors: Methodologies and Applications.J Chem Theory Comput. 2023 Dec 12;19(23):8491-8522. doi: 10.1021/acs.jctc.3c00960. Epub 2023 Nov 20. J Chem Theory Comput. 2023. PMID: 37984502 Review.
-
Generalized Semiclassical Ehrenfest Method: A Route to Wave Function-Free Photochemistry and Nonadiabatic Dynamics with Only Potential Energies and Gradients.J Chem Theory Comput. 2024 Jun 11;20(11):4396-4426. doi: 10.1021/acs.jctc.4c00424. Epub 2024 May 31. J Chem Theory Comput. 2024. PMID: 38819014 Review.
Cited by
-
Monitoring the Evolution of Relative Product Populations at Early Times during a Photochemical Reaction.J Am Chem Soc. 2024 Feb 14;146(6):4134-4143. doi: 10.1021/jacs.3c13046. Epub 2024 Feb 5. J Am Chem Soc. 2024. PMID: 38317439 Free PMC article.
-
Molecular structure retrieval directly from laboratory-frame photoelectron spectra in laser-induced electron diffraction.Nat Commun. 2021 Mar 9;12(1):1520. doi: 10.1038/s41467-021-21855-4. Nat Commun. 2021. PMID: 33750798 Free PMC article.
-
Computational and experimental investigation of the effect of cation structure on the solubility of anionic flow battery active-materials.Chem Sci. 2021 Nov 26;12(48):15892-15907. doi: 10.1039/d1sc04990a. eCollection 2021 Dec 15. Chem Sci. 2021. PMID: 35024113 Free PMC article.
-
Excited-State Dynamics Simulations of a Light-Driven Molecular Motor in Solution.J Phys Chem A. 2023 Nov 16;127(45):9520-9529. doi: 10.1021/acs.jpca.3c05841. Epub 2023 Nov 2. J Phys Chem A. 2023. PMID: 37917883 Free PMC article.
-
PyUNIxMD: A Python-based excited state molecular dynamics package.J Comput Chem. 2021 Sep 15;42(24):1755-1766. doi: 10.1002/jcc.26711. Epub 2021 Jul 1. J Comput Chem. 2021. PMID: 34197646 Free PMC article.
References
FURTHER READING
-
- The SHARC2.0 package and the WFOVERLAP program, as well as comprehensive documentation and tutorials, can be obtained at http://sharc-md.org/
References
-
- Amatatsu, Y. , Morokuma, K. , & Yabushita, S. (1991). Ab initio potential energy surfaces and trajectory studies of A‐band photodissociation dynamics: CH3I*→ CH3 + I and CH3 + I* . The Journal of Chemical Physics, 94(7), 4858–4876. 10.1063/1.460571 - DOI
Publication types
LinkOut - more resources
Full Text Sources
Research Materials