

Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement
- PMID: 30451828
- PMCID: PMC6242939
- DOI: 10.1038/s41467-018-07307-6
Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement
Abstract
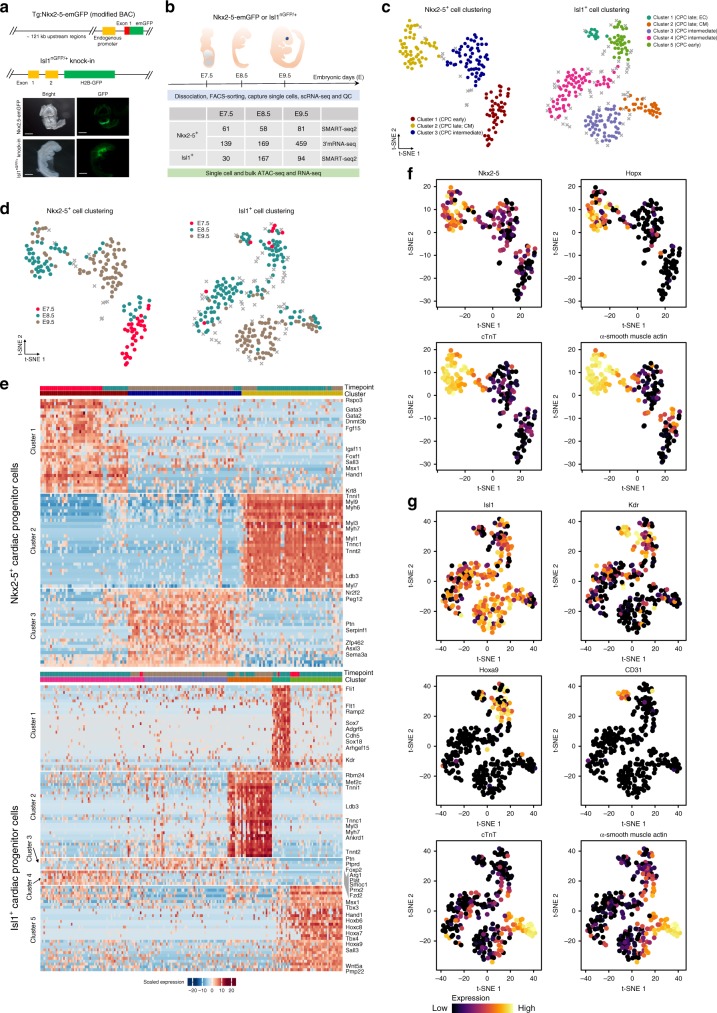
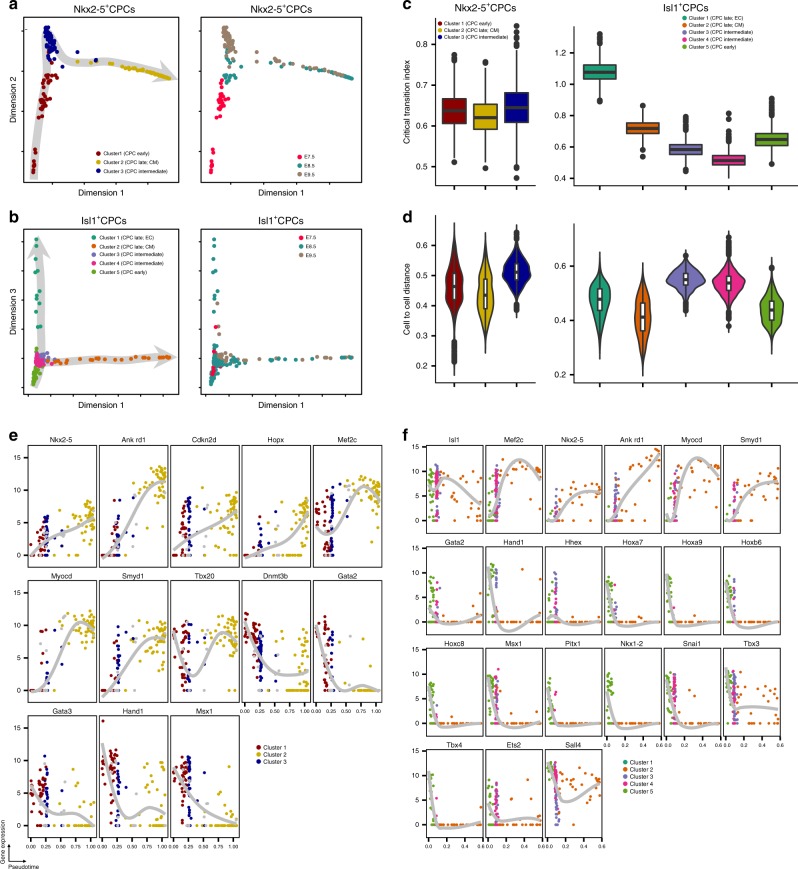
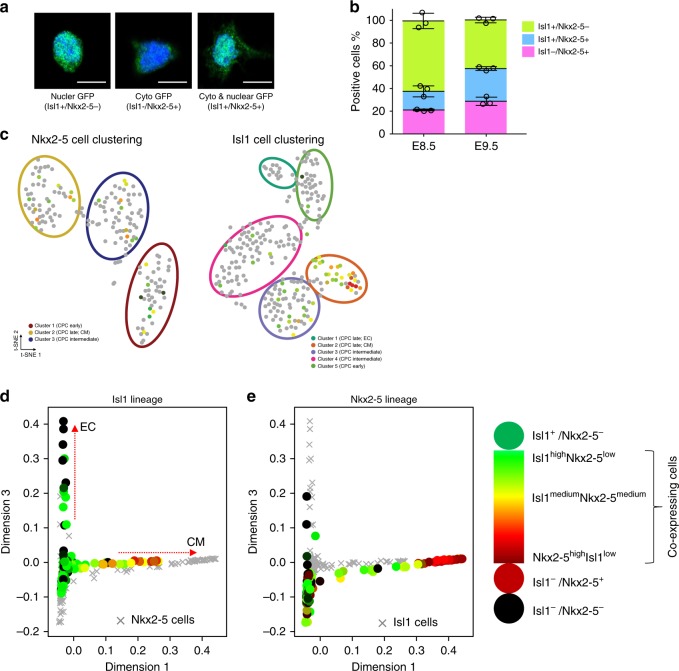
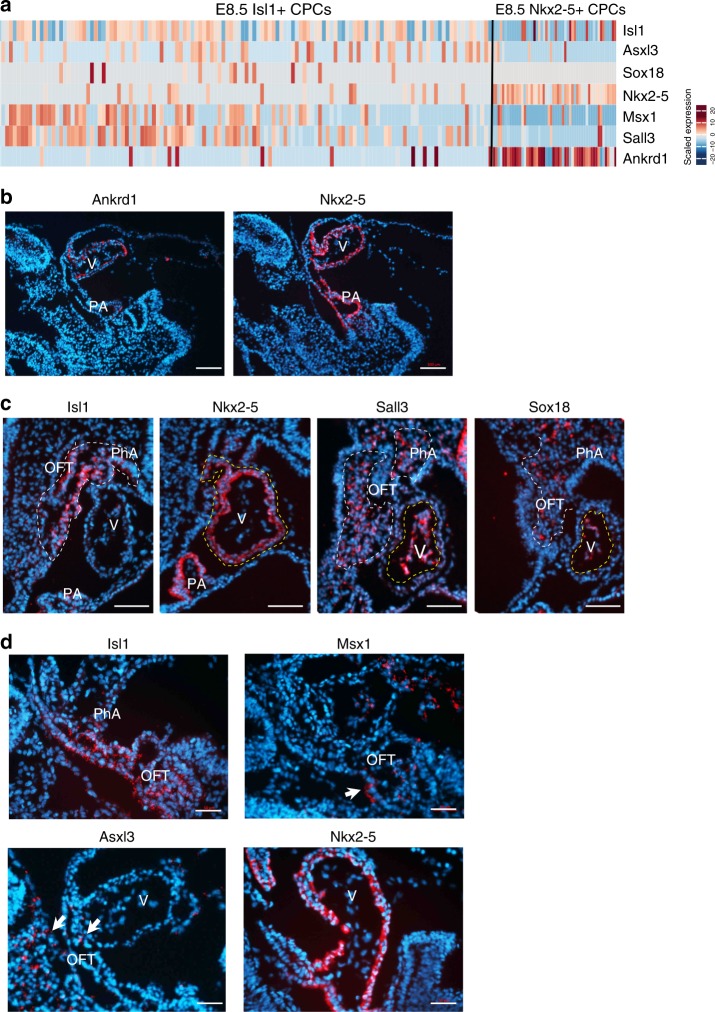
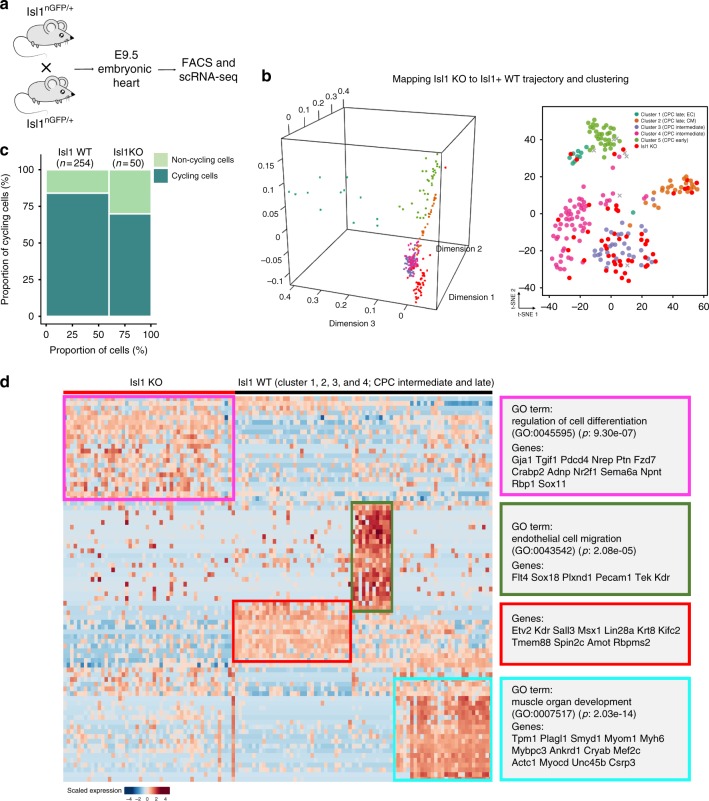
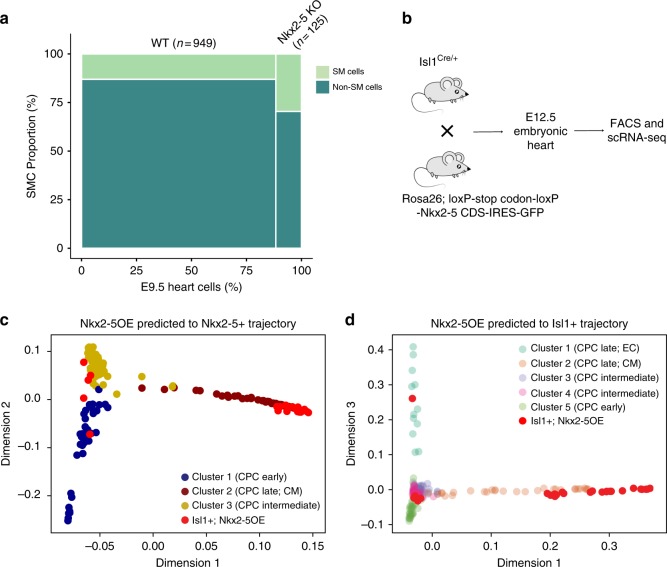
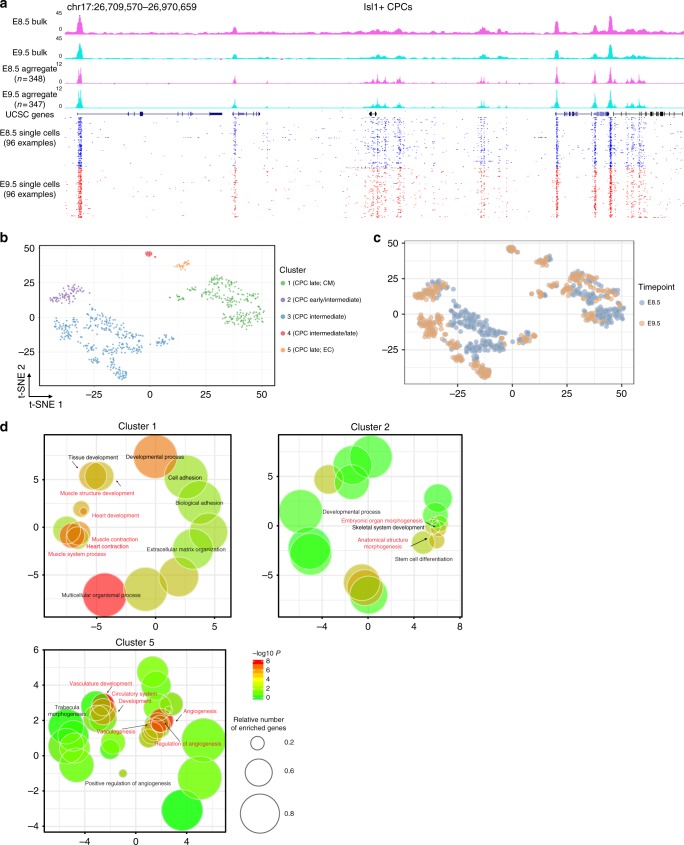
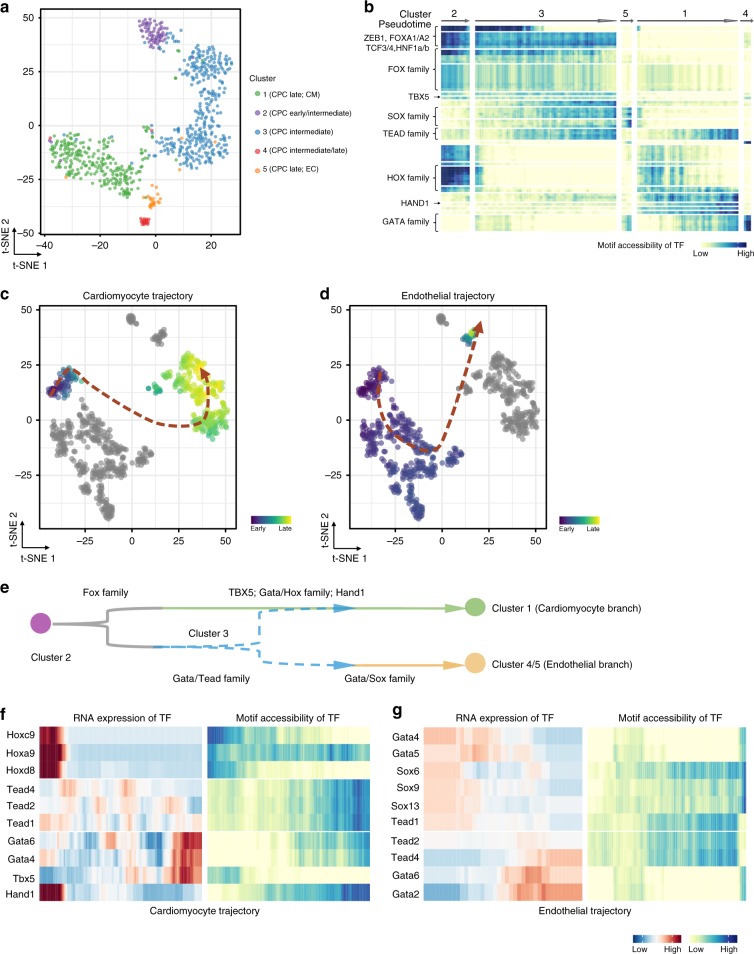
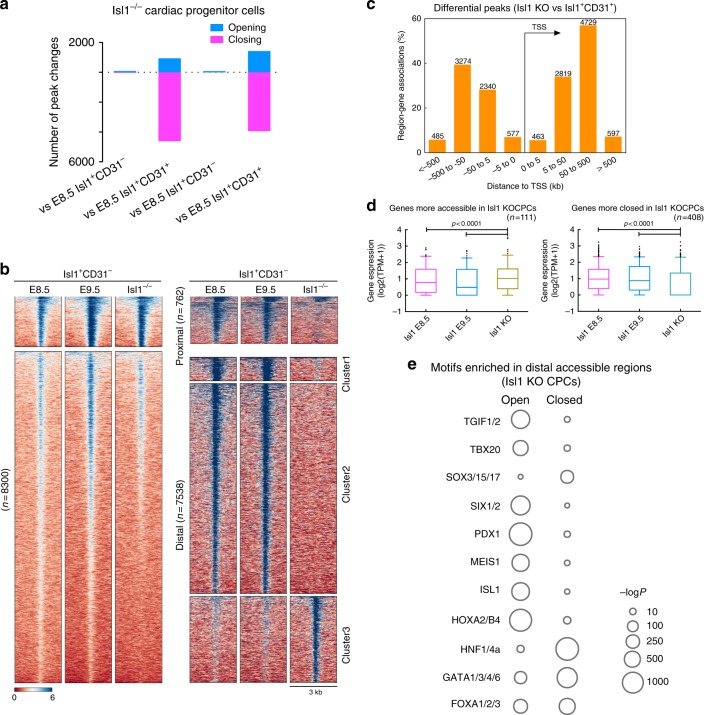
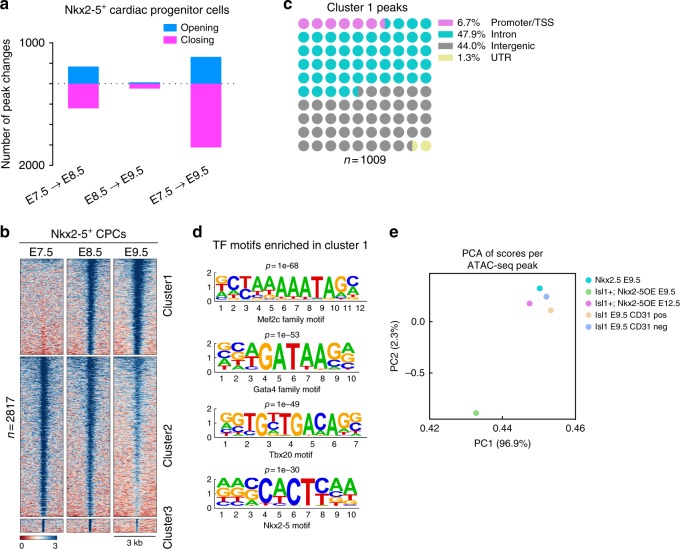
Formation and segregation of cell lineages forming the heart have been studied extensively but the underlying gene regulatory networks and epigenetic changes driving cell fate transitions during early cardiogenesis are still only partially understood. Here, we comprehensively characterize mouse cardiac progenitor cells (CPCs) marked by Nkx2-5 and Isl1 expression from E7.5 to E9.5 using single-cell RNA sequencing and transposase-accessible chromatin profiling (ATAC-seq). By leveraging on cell-to-cell transcriptome and chromatin accessibility heterogeneity, we identify different previously unknown cardiac subpopulations. Reconstruction of developmental trajectories reveal that multipotent Isl1+ CPC pass through an attractor state before separating into different developmental branches, whereas extended expression of Nkx2-5 commits CPC to an unidirectional cardiomyocyte fate. Furthermore, we show that CPC fate transitions are associated with distinct open chromatin states critically depending on Isl1 and Nkx2-5. Our data provide a model of transcriptional and epigenetic regulations during cardiac progenitor cell fate decisions at single-cell resolution.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
