

POLG-related disorders and their neurological manifestations
- PMID: 30451971
- PMCID: PMC8796686
- DOI: 10.1038/s41582-018-0101-0
POLG-related disorders and their neurological manifestations
Abstract
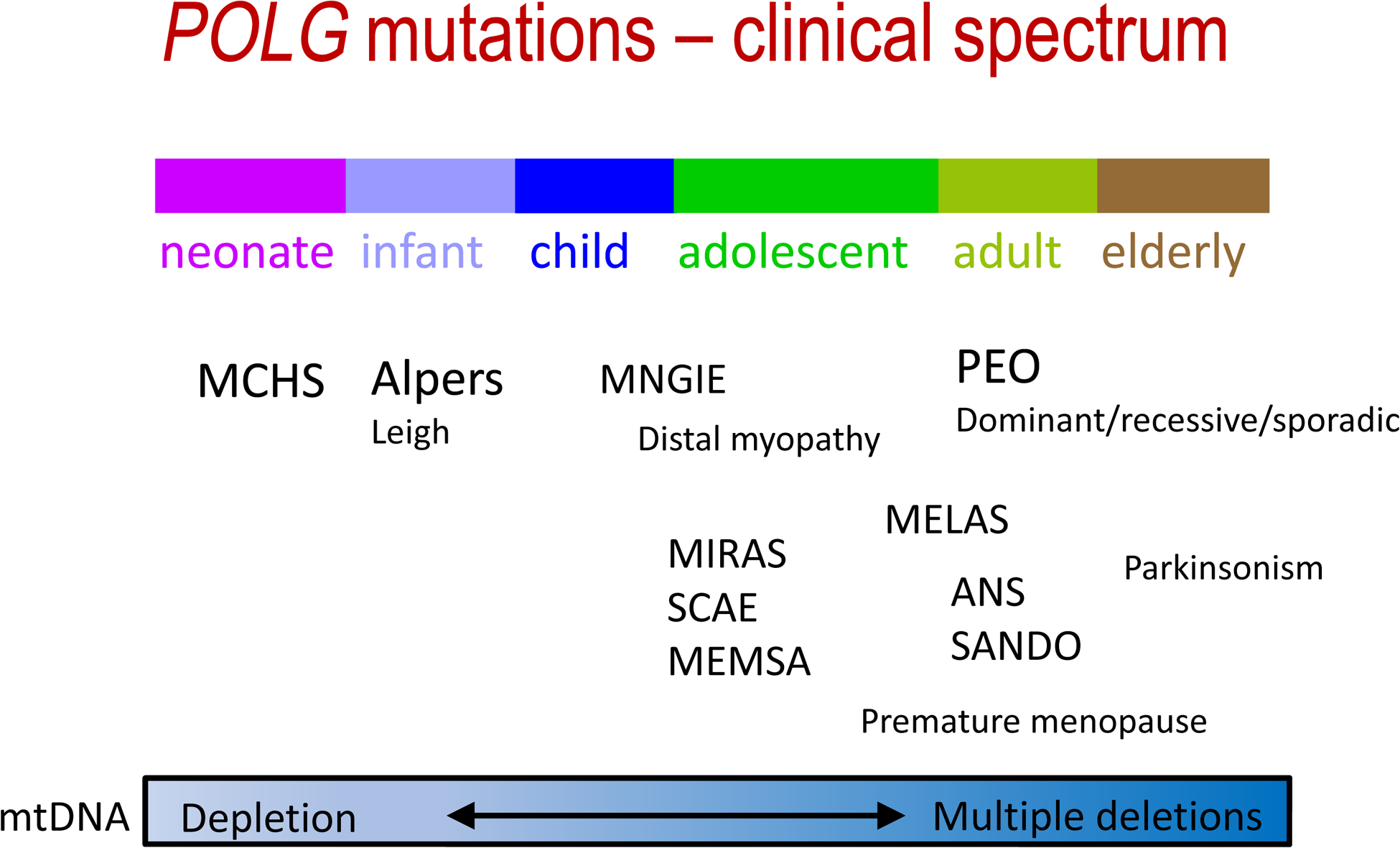
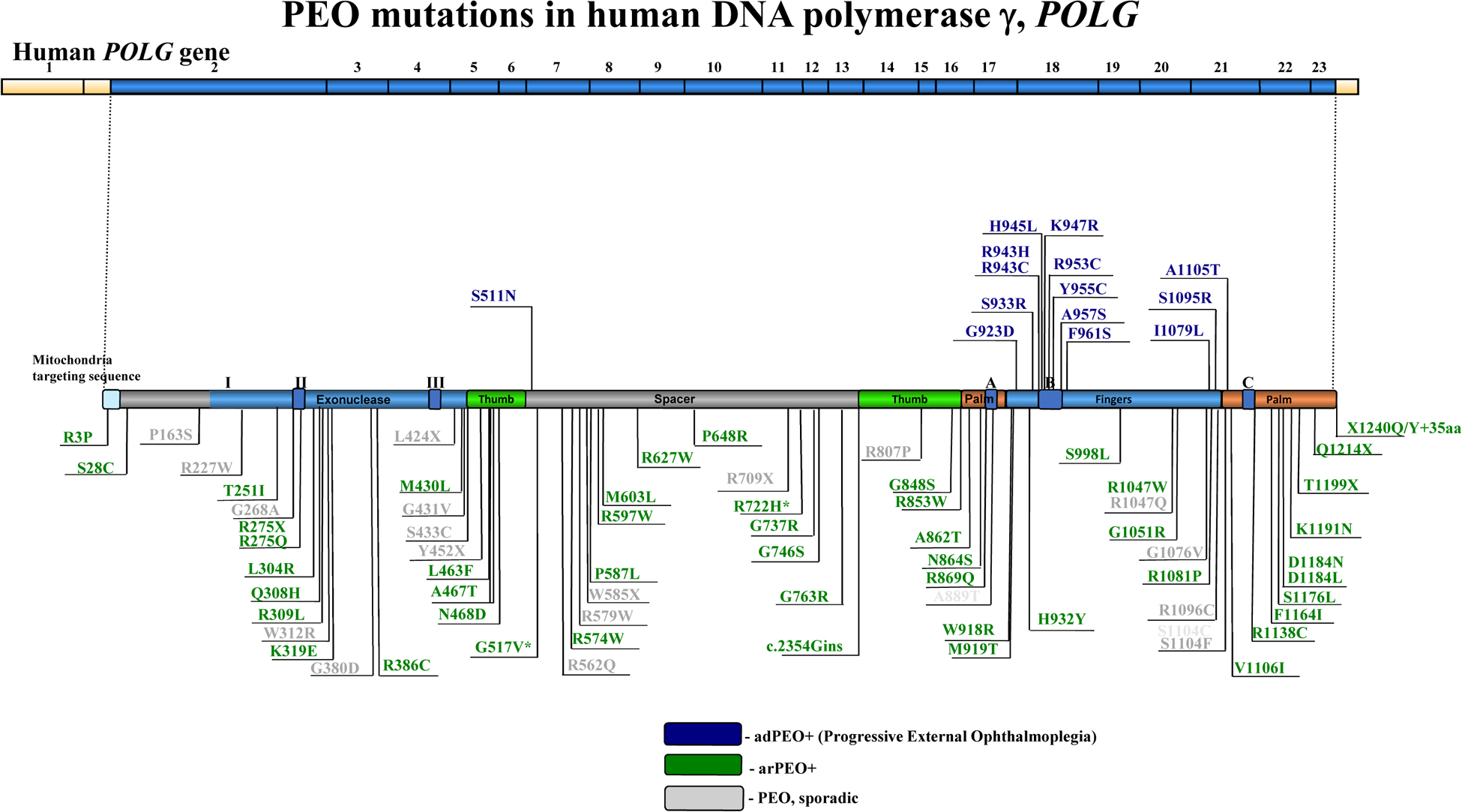
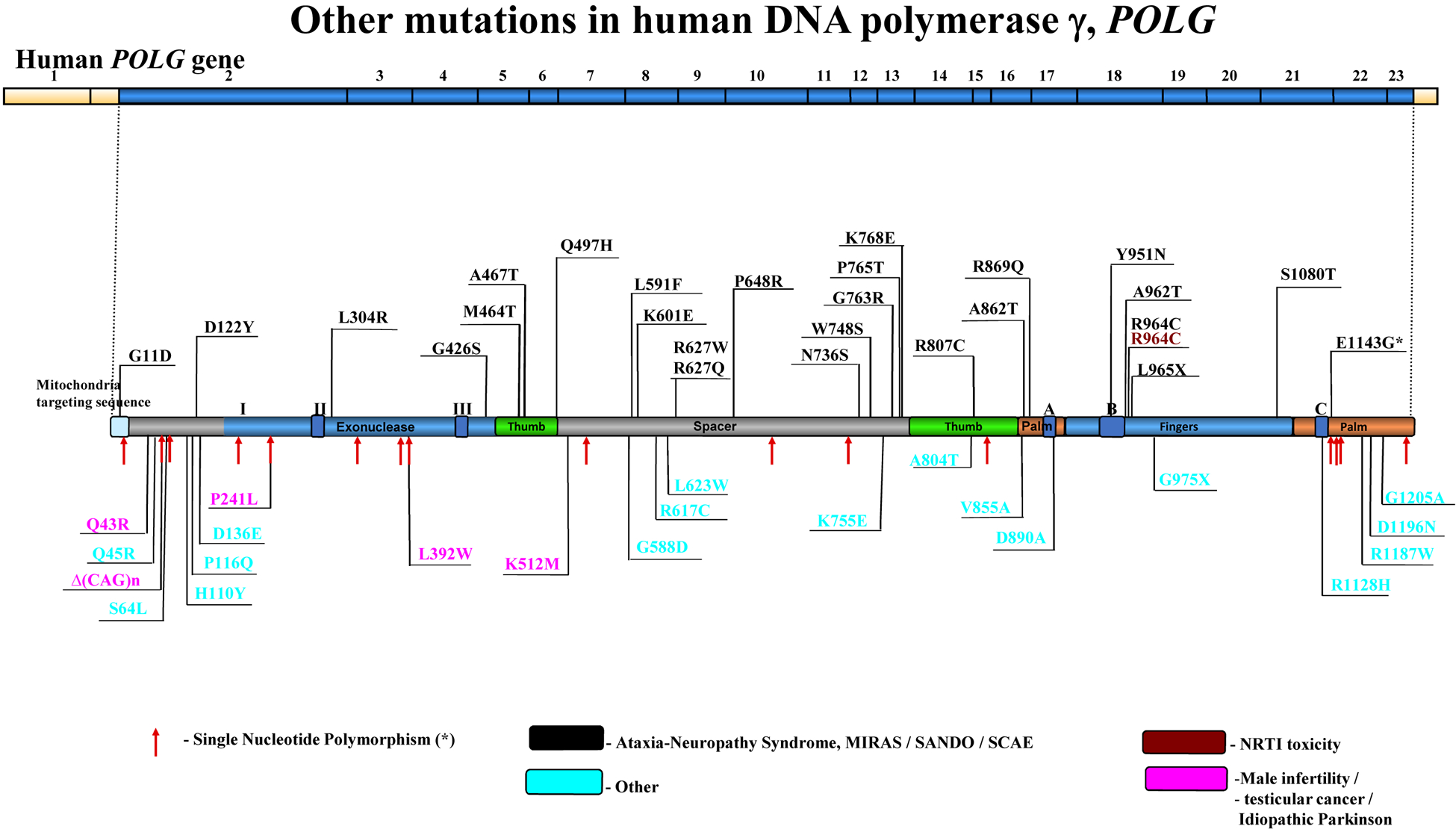
The POLG gene encodes the mitochondrial DNA polymerase that is responsible for replication of the mitochondrial genome. Mutations in POLG can cause early childhood mitochondrial DNA (mtDNA) depletion syndromes or later-onset syndromes arising from mtDNA deletions. POLG mutations are the most common cause of inherited mitochondrial disorders, with as many as 2% of the population carrying these mutations. POLG-related disorders comprise a continuum of overlapping phenotypes with onset from infancy to late adulthood. The six leading disorders caused by POLG mutations are Alpers-Huttenlocher syndrome, which is one of the most severe phenotypes; childhood myocerebrohepatopathy spectrum, which presents within the first 3 years of life; myoclonic epilepsy myopathy sensory ataxia; ataxia neuropathy spectrum; autosomal recessive progressive external ophthalmoplegia; and autosomal dominant progressive external ophthalmoplegia. This Review describes the clinical features, pathophysiology, natural history and treatment of POLG-related disorders, focusing particularly on the neurological manifestations of these conditions.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
