

Evolutionary dynamics of residual disease in human glioblastoma
- PMID: 30452544
- PMCID: PMC6442656
- DOI: 10.1093/annonc/mdy506
Evolutionary dynamics of residual disease in human glioblastoma
Abstract
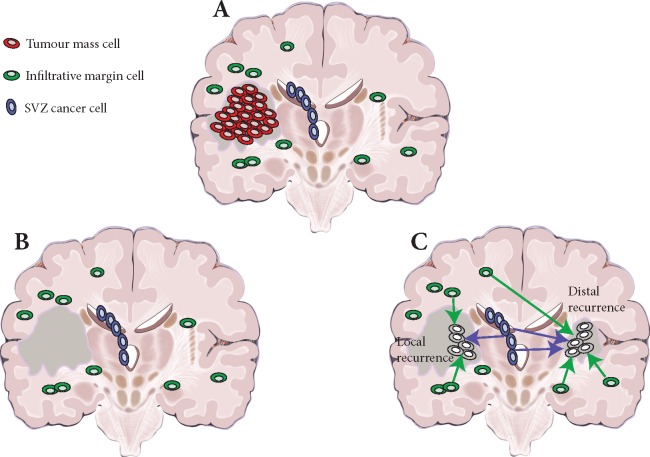
Background: Glioblastoma is the most common and aggressive adult brain malignancy against which conventional surgery and chemoradiation provide limited benefit. Even when a good treatment response is obtained, recurrence inevitably occurs either locally (∼80%) or distally (∼20%), driven by cancer clones that are often genomically distinct from those in the primary tumour. Glioblastoma cells display a characteristic infiltrative phenotype, invading the surrounding tissue and often spreading across the whole brain. Cancer cells responsible for relapse can reside in two compartments of residual disease that are left behind after treatment: the infiltrated normal brain parenchyma and the sub-ventricular zone. However, these two sources of residual disease in glioblastoma are understudied because of the difficulty in sampling these regions during surgery.
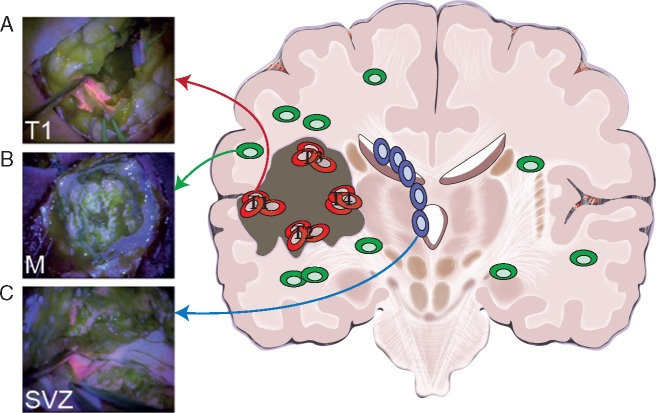
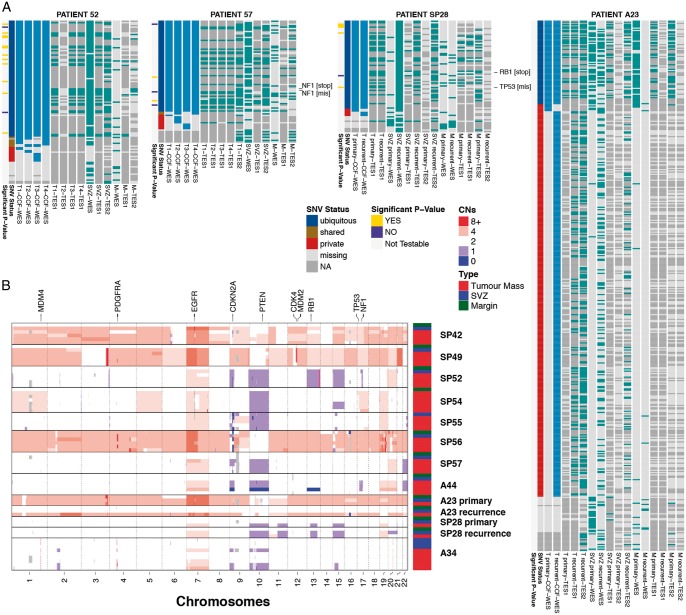
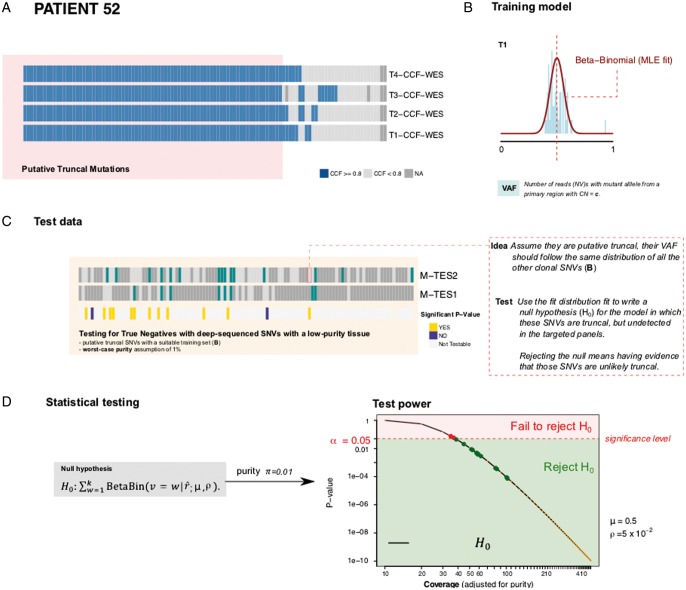
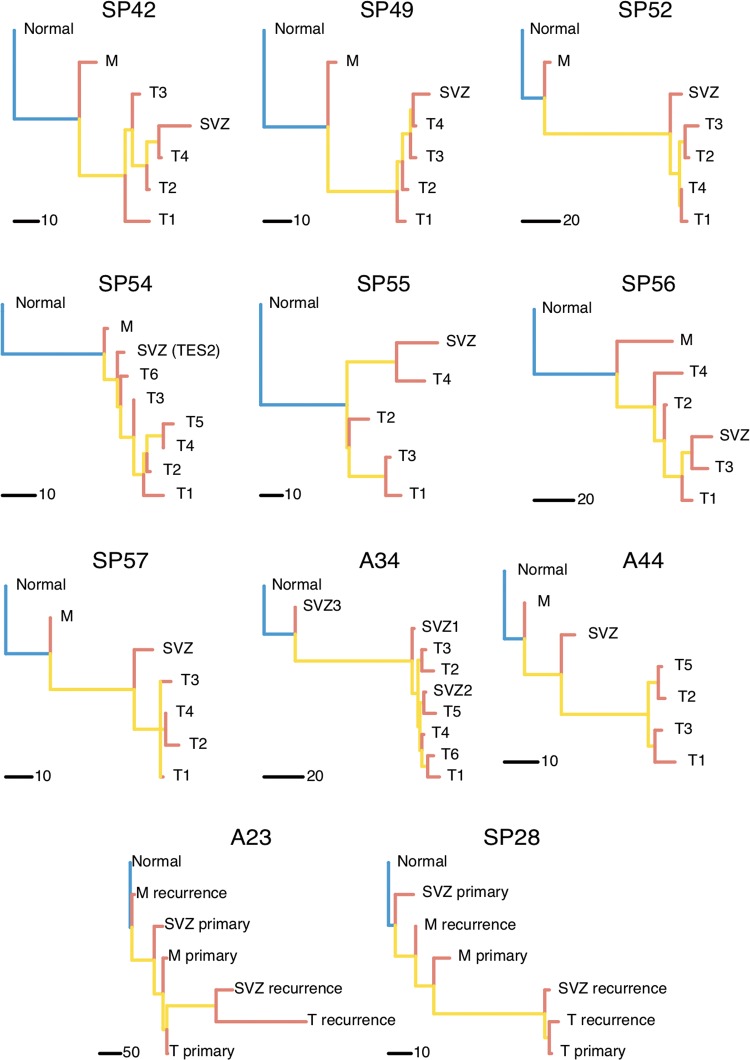
Patient and methods: Here, we present the results of whole-exome sequencing of 69 multi-region samples collected using fluorescence-guided resection from 11 patients, including the infiltrating tumour margin and the sub-ventricular zone for each patient, as well as matched blood. We used a phylogenomic approach to dissect the spatio-temporal evolution of each tumour and unveil the relation between residual disease and the main tumour mass. We also analysed two patients with paired primary-recurrence samples with matched residual disease.
Results: Our results suggest that infiltrative subclones can arise early during tumour growth in a subset of patients. After treatment, the infiltrative subclones may seed the growth of a recurrent tumour, thus representing the 'missing link' between the primary tumour and recurrent disease.
Conclusions: These results are consistent with recognised clinical phenotypic behaviour and suggest that more specific therapeutic targeting of cells in the infiltrated brain parenchyma may improve patient's outcome.
Keywords: cancer evolution; glioblastoma; phylogenetics; sub-ventricular zone; tumour margin.
© The Author(s) 2018. Published by Oxford University Press on behalf of the European Society for Medical Oncology.
Figures
Comment in
-
Searching for the needle in the haystack: deconvoluting the evolutionary dynamics of residual disease in human glioblastoma.Ann Oncol. 2019 Mar 1;30(3):355-357. doi: 10.1093/annonc/mdz042. Ann Oncol. 2019. PMID: 30753265 Free PMC article. No abstract available.
References
-
- Stupp R, Mason WP, van den Bent MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352(10): 987–996. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
