

Mutations of the mitochondrial carrier translocase channel subunit TIM22 cause early-onset mitochondrial myopathy
- PMID: 30452684
- PMCID: PMC6240735
- DOI: 10.1093/hmg/ddy305
Mutations of the mitochondrial carrier translocase channel subunit TIM22 cause early-onset mitochondrial myopathy
Abstract
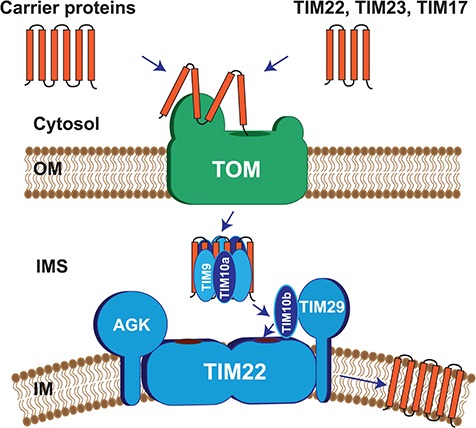
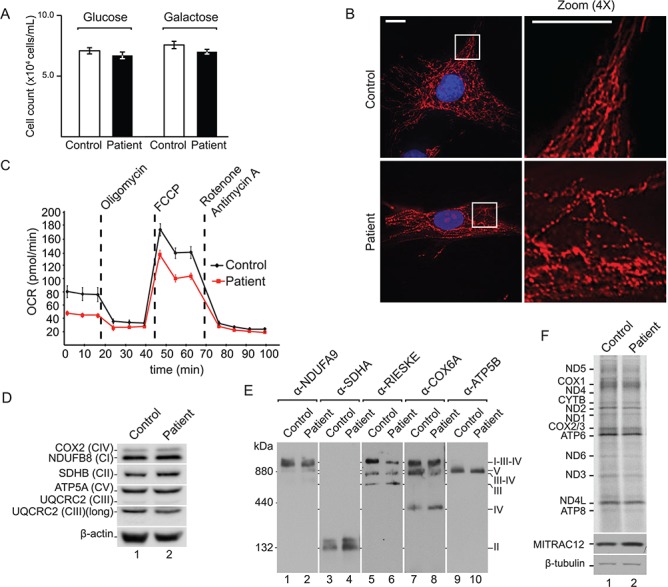
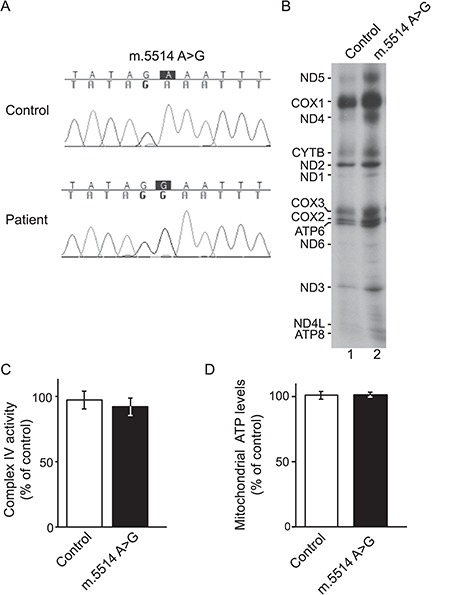
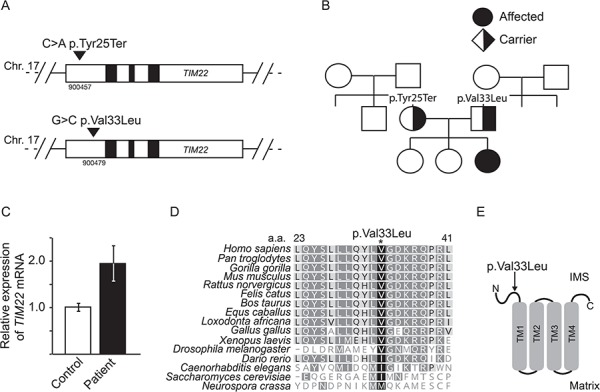
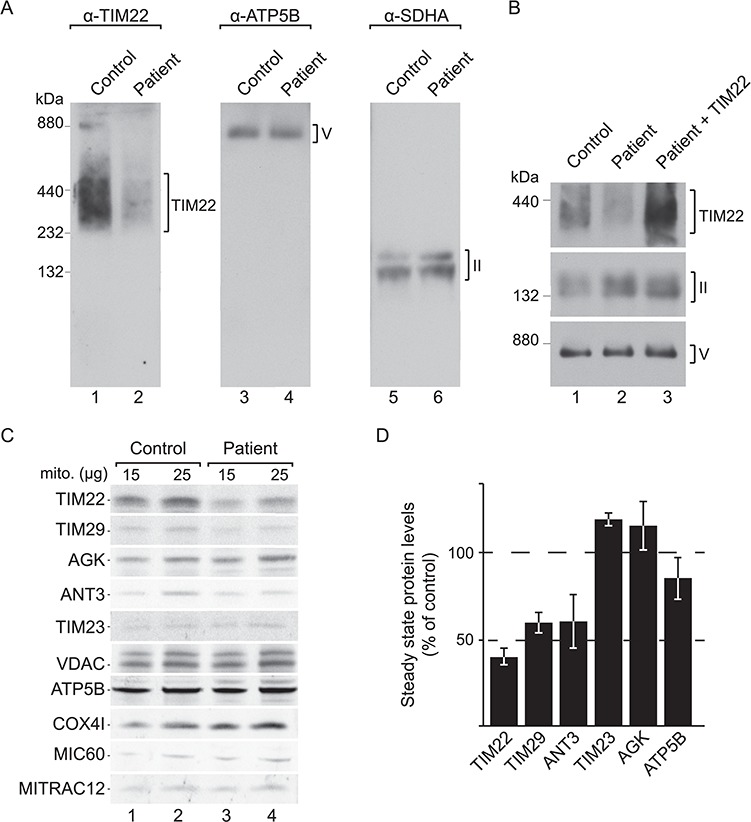
Protein import into mitochondria is facilitated by translocases within the outer and the inner mitochondrial membranes that are dedicated to a highly specific subset of client proteins. The mitochondrial carrier translocase (TIM22 complex) inserts multispanning proteins, such as mitochondrial metabolite carriers and translocase subunits (TIM23, TIM17A/B and TIM22), into the inner mitochondrial membrane. Both types of substrates are essential for mitochondrial metabolic function and biogenesis. Here, we report on a subject, diagnosed at 1.5 years, with a neuromuscular presentation, comprising hypotonia, gastroesophageal reflux disease and persistently elevated serum and Cerebrospinal fluid lactate (CSF). Patient fibroblasts displayed reduced oxidative capacity and altered mitochondrial morphology. Using trans-mitochondrial cybrid cell lines, we excluded a candidate variant in mitochondrial DNA as causative of these effects. Whole-exome sequencing identified compound heterozygous variants in the TIM22 gene (NM_013337), resulting in premature truncation in one allele (p.Tyr25Ter) and a point mutation in a conserved residue (p.Val33Leu), within the intermembrane space region, of the TIM22 protein in the second allele. Although mRNA transcripts of TIM22 were elevated, biochemical analyses revealed lower levels of TIM22 protein and an even greater deficiency of TIM22 complex formation. In agreement with a defect in carrier translocase function, carrier protein amounts in the inner membrane were found to be reduced. This is the first report of pathogenic variants in the TIM22 pore-forming subunit of the carrier translocase affecting the biogenesis of inner mitochondrial membrane proteins critical for metabolite exchange.
Figures
References
-
- Dudek J., Rehling P. and Laan M. (2013) Mitochondrial protein import; common principles and physiological networks. Biochim. Biophys. Acta, 1833, 274–285. - PubMed
-
- Wiedemann N. and Pfanner N. (2017) Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem., 86, 685–714. - PubMed
-
- Endo T., Yamano K. and Kawano S. (2011) Structural insight into the mitochondrial protein import system. Biochim. Biophys. Acta, 1808, 955–970. - PubMed
-
- Neupert W. and Herrmann J.M. (2007) Translocation of proteins into mitochondria. Annu. Rev. Biochem., 76, 723–749. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
