

The physiology of foamy phagocytes in multiple sclerosis
- PMID: 30454040
- PMCID: PMC6240956
- DOI: 10.1186/s40478-018-0628-8
The physiology of foamy phagocytes in multiple sclerosis
Abstract
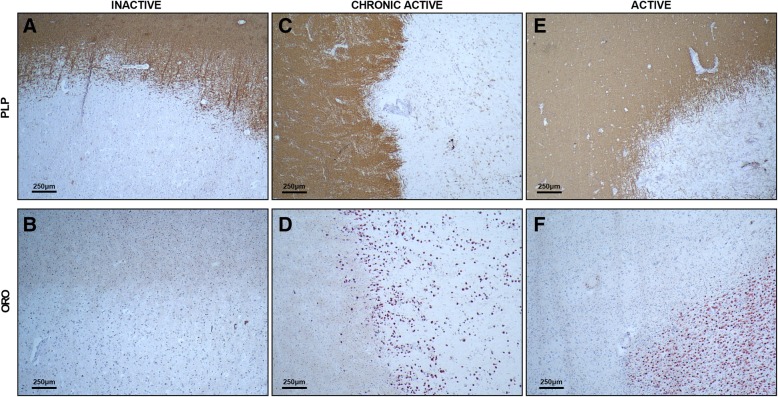
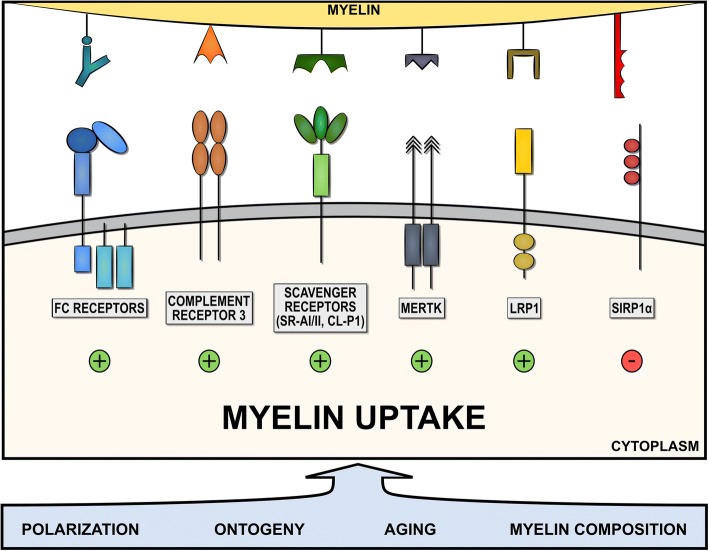
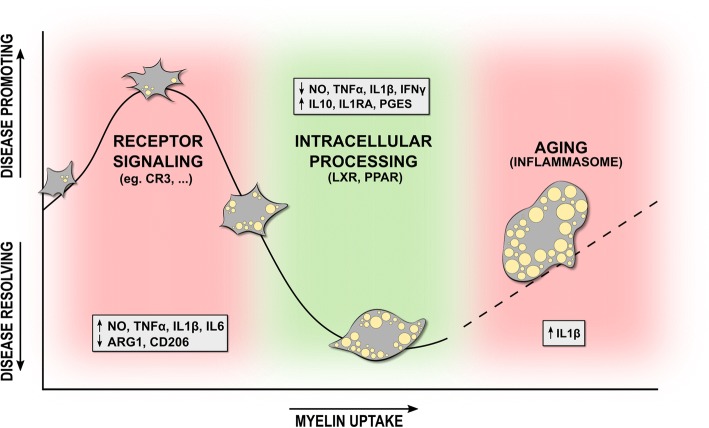
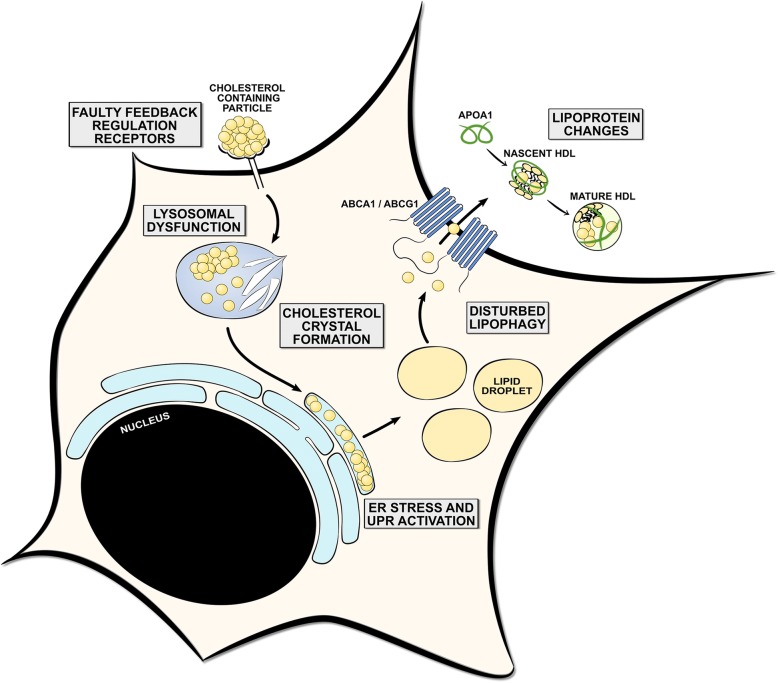
Multiple sclerosis (MS) is a chronic disease of the central nervous system characterized by massive infiltration of immune cells, demyelination, and axonal loss. Active MS lesions mainly consist of macrophages and microglia containing abundant intracellular myelin remnants. Initial studies showed that these foamy phagocytes primarily promote MS disease progression by internalizing myelin debris, presenting brain-derived autoantigens, and adopting an inflammatory phenotype. However, more recent studies indicate that phagocytes can also adopt a beneficial phenotype upon myelin internalization. In this review, we summarize and discuss the current knowledge on the spatiotemporal physiology of foamy phagocytes in MS lesions, and elaborate on extrinsic and intrinsic factors regulating their behavior. In addition, we discuss and link the physiology of myelin-containing phagocytes to that of foamy macrophages in other disorders such atherosclerosis.
Keywords: Macrophage; Microglia; Multiple sclerosis; Neuroinflammation; Polarization; Remyelination.
Conflict of interest statement
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
