

Physiological and pathophysiological characteristics of ataxin-3 isoforms
- PMID: 30455355
- PMCID: PMC6333899
- DOI: 10.1074/jbc.RA118.005801
Physiological and pathophysiological characteristics of ataxin-3 isoforms
Abstract
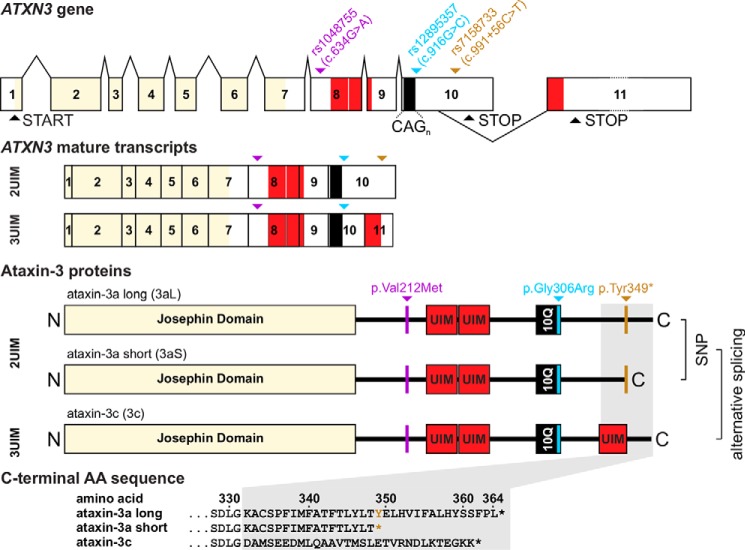
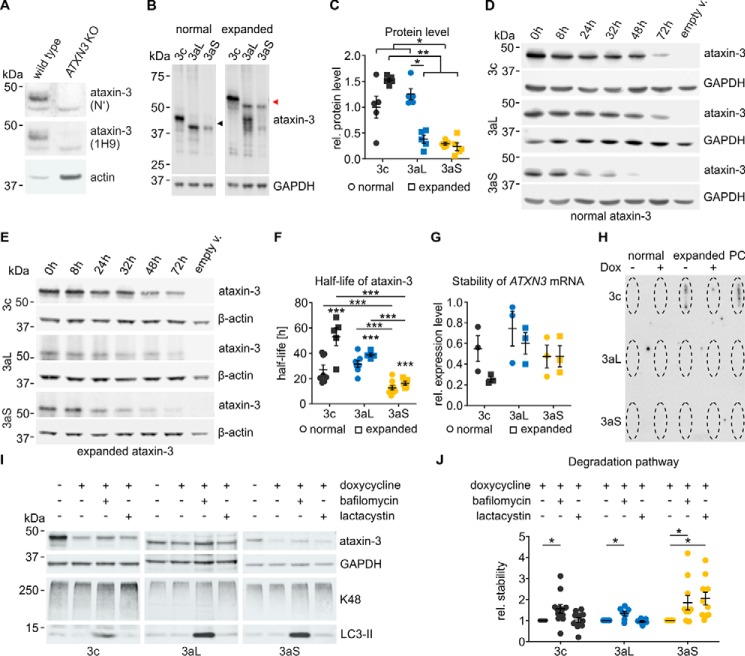
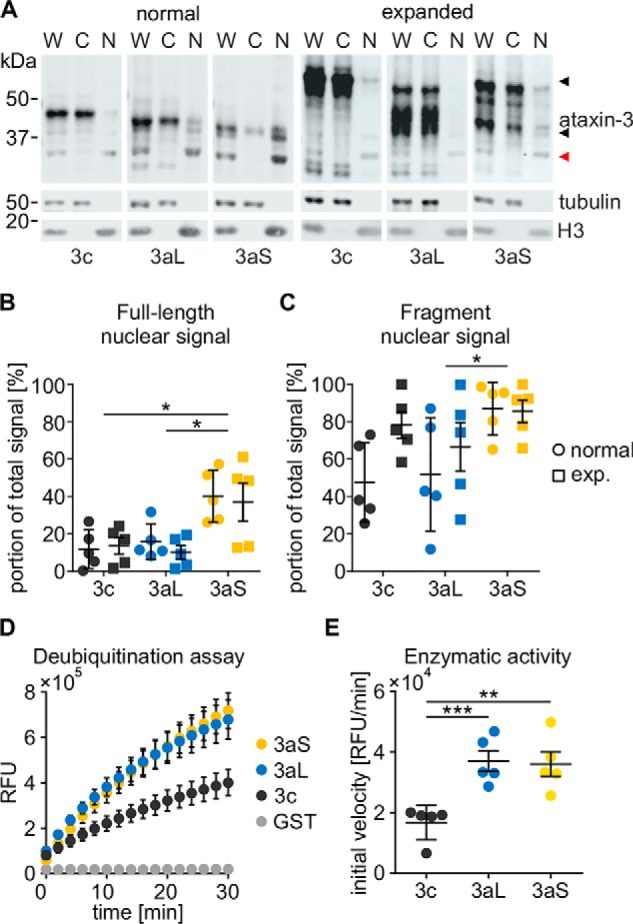
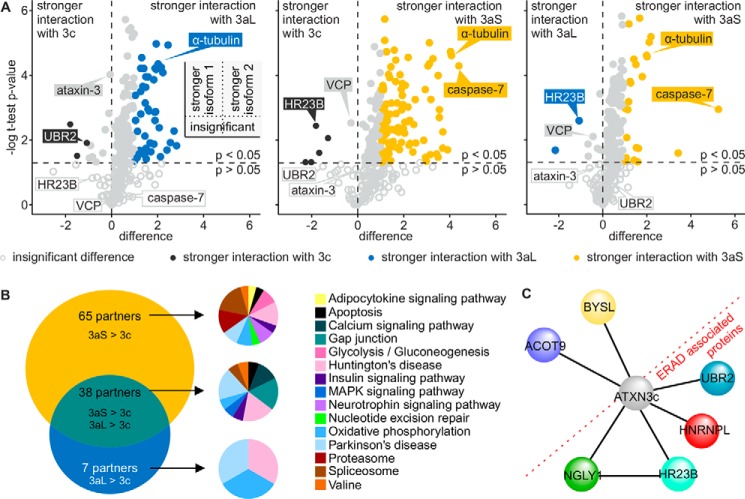
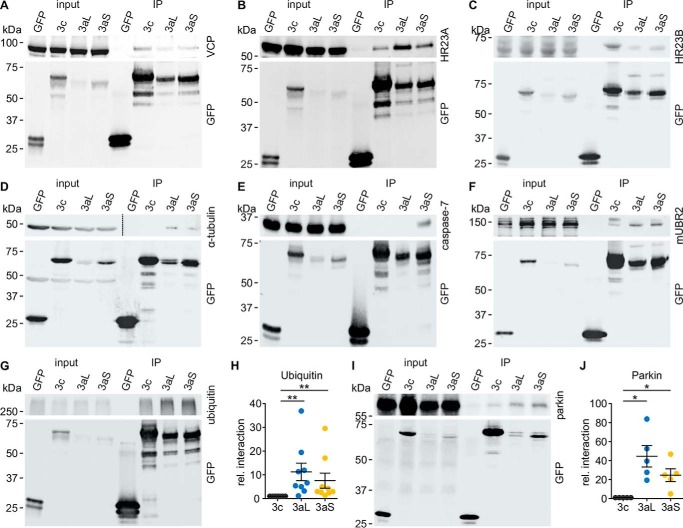
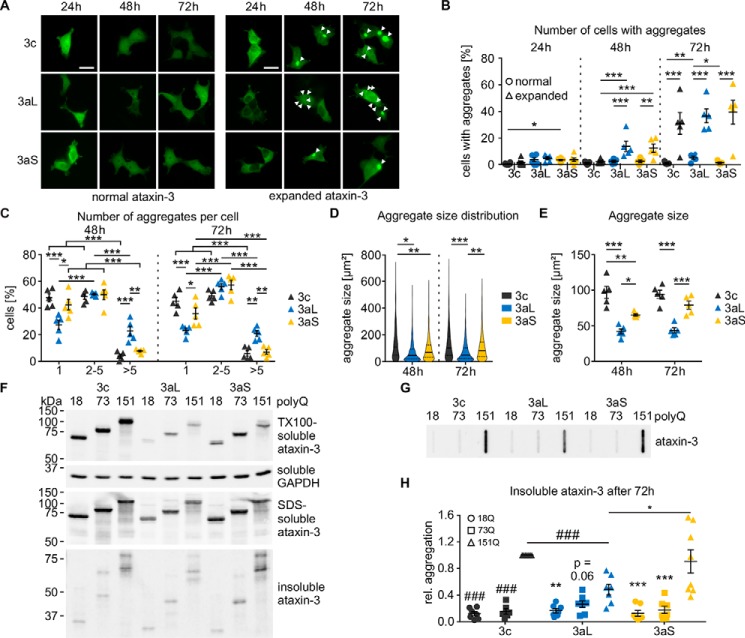
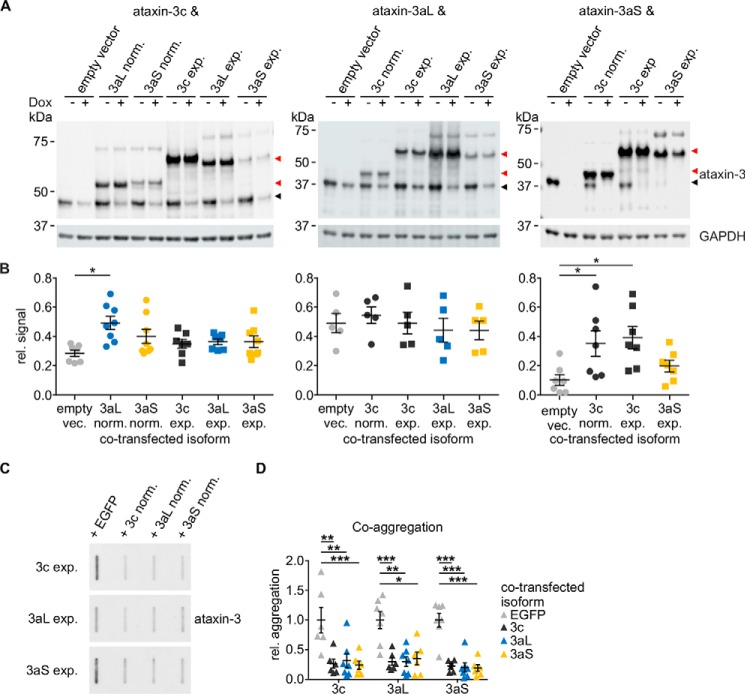
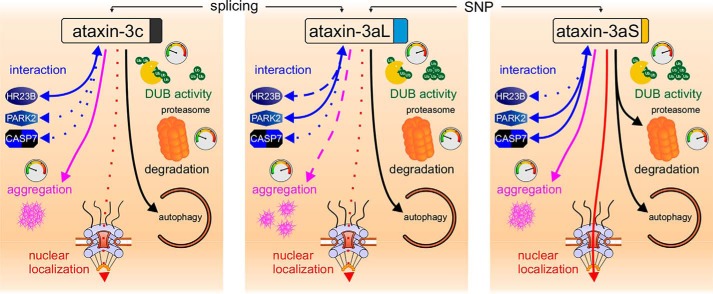
Ataxin-3 is a deubiquitinating enzyme and the affected protein in the neurodegenerative disorder Machado-Joseph disease (MJD). The ATXN3 gene is alternatively spliced, resulting in protein isoforms that differ in the number of ubiquitin-interacting motifs. Additionally, nonsynonymous SNPs in ATXN3 cause amino acid changes in ataxin-3, and one of these polymorphisms introduces a premature stop codon in one isoform. Here, we examined the effects of different ataxin-3 isoforms and of the premature stop codon on ataxin-3's physiological function and on main disease mechanisms. At the physiological level, we show that alternative splicing and the premature stop codon alter ataxin-3 stability and that ataxin-3 isoforms differ in their enzymatic deubiquitination activity, subcellular distribution, and interaction with other proteins. At the pathological level, we found that the expansion of the polyglutamine repeat leads to a stabilization of ataxin-3 and that ataxin-3 isoforms differ in their aggregation properties. Interestingly, we observed a functional interaction between normal and polyglutamine-expanded ATXN3 allelic variants. We found that interactions between different ATXN3 allelic variants modify the physiological and pathophysiological properties of ataxin-3. Our findings indicate that alternative splicing and interactions between different ataxin-3 isoforms affect not only major aspects of ataxin-3 function but also MJD pathogenesis. Our results stress the importance of considering isoforms of disease-causing proteins and their interplay with the normal allelic variant as disease modifiers in MJD and autosomal-dominantly inherited diseases in general.
Keywords: E3 ubiquitin ligase; alternative splicing; deubiquitylation (deubiquitination); enzyme degradation; enzyme kinetics; genetic polymorphism; neurodegenerative disease; polyglutamine disease; protein aggregation; proteomics.
© 2019 Weishäupl et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Schmidt T., Landwehrmeyer G. B., Schmitt I., Trottier Y., Auburger G., Laccone F., Klockgether T., Völpel M., Epplen J. T., Schöls L., and Riess O. (1998) An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 8, 669–679 10.1111/j.1750-3639.1998.tb00193.x - DOI - PMC - PubMed
-
- Ichikawa Y., Goto J., Hattori M., Toyoda A., Ishii K., Jeong S. Y., Hashida H., Masuda N., Ogata K., Kasai F., Hirai M., Maciel P., Rouleau G. A., Sakaki Y., and Kanazawa I. (2001) The genomic structure and expression of MJD, the Machado–Joseph disease gene. J. Hum. Genet. 46, 413–422 10.1007/s100380170060 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
