

The Interplay Between Immune Response and Bacterial Infection in COPD: Focus Upon Non-typeable Haemophilus influenzae
- PMID: 30455693
- PMCID: PMC6230626
- DOI: 10.3389/fimmu.2018.02530
The Interplay Between Immune Response and Bacterial Infection in COPD: Focus Upon Non-typeable Haemophilus influenzae
Abstract
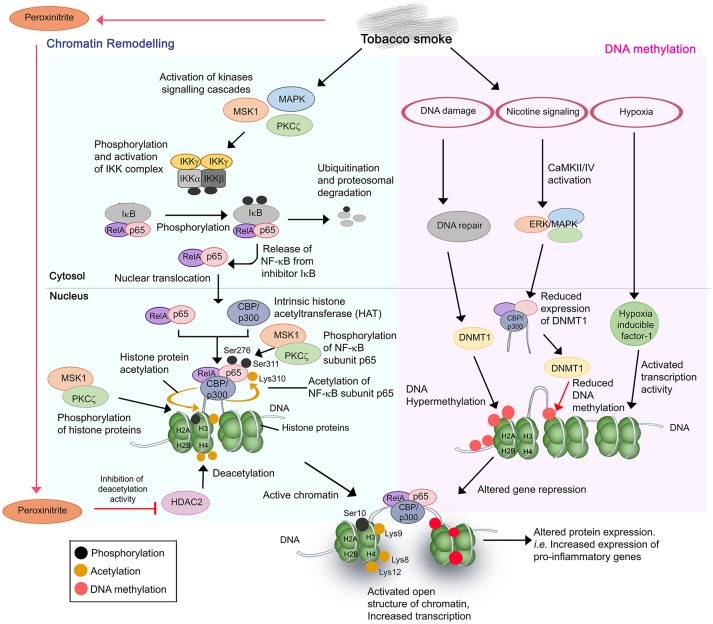
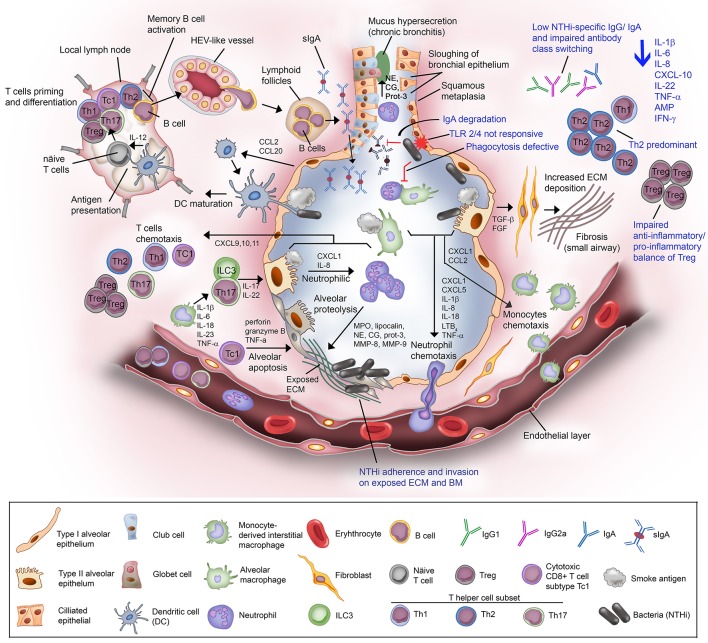
Chronic obstructive pulmonary disease (COPD) is a debilitating respiratory disease and one of the leading causes of morbidity and mortality worldwide. It is characterized by persistent respiratory symptoms and airflow limitation due to abnormalities in the lower airway following consistent exposure to noxious particles or gases. Acute exacerbations of COPD (AECOPD) are characterized by increased cough, purulent sputum production, and dyspnea. The AECOPD is mostly associated with infection caused by common cold viruses or bacteria, or co-infections. Chronic and persistent infection by non-typeable Haemophilus influenzae (NTHi), a Gram-negative coccobacillus, contributes to almost half of the infective exacerbations caused by bacteria. This is supported by reports that NTHi is commonly isolated in the sputum from COPD patients during exacerbations. Persistent colonization of NTHi in the lower airway requires a plethora of phenotypic adaptation and virulent mechanisms that are developed over time to cope with changing environmental pressures in the airway such as host immuno-inflammatory response. Chronic inhalation of noxious irritants in COPD causes a changed balance in the lung microbiome, abnormal inflammatory response, and an impaired airway immune system. These conditions significantly provide an opportunistic platform for NTHi colonization and infection resulting in a "vicious circle." Episodes of large inflammation as the consequences of multiple interactions between airway immune cells and NTHi, accumulatively contribute to COPD exacerbations and may result in worsening of the clinical status. In this review, we discuss in detail the interplay and crosstalk between airway immune residents and NTHi, and their effect in AECOPD for better understanding of NTHi pathogenesis in COPD patients.
Keywords: COPD; airway; exacerbation; immune response; infection; inflammation; non-typeable Haemophilus influenza.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical