

mTORC1 is a key mediator of RON-dependent breast cancer metastasis with therapeutic potential
- PMID: 30456298
- PMCID: PMC6226524
- DOI: 10.1038/s41523-018-0091-5
mTORC1 is a key mediator of RON-dependent breast cancer metastasis with therapeutic potential
Abstract
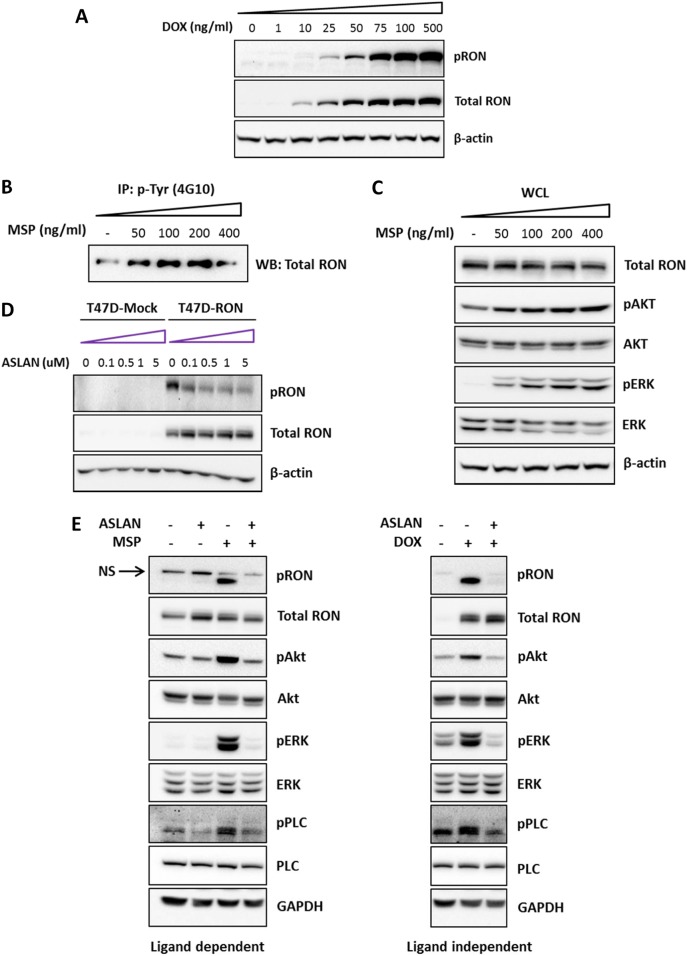
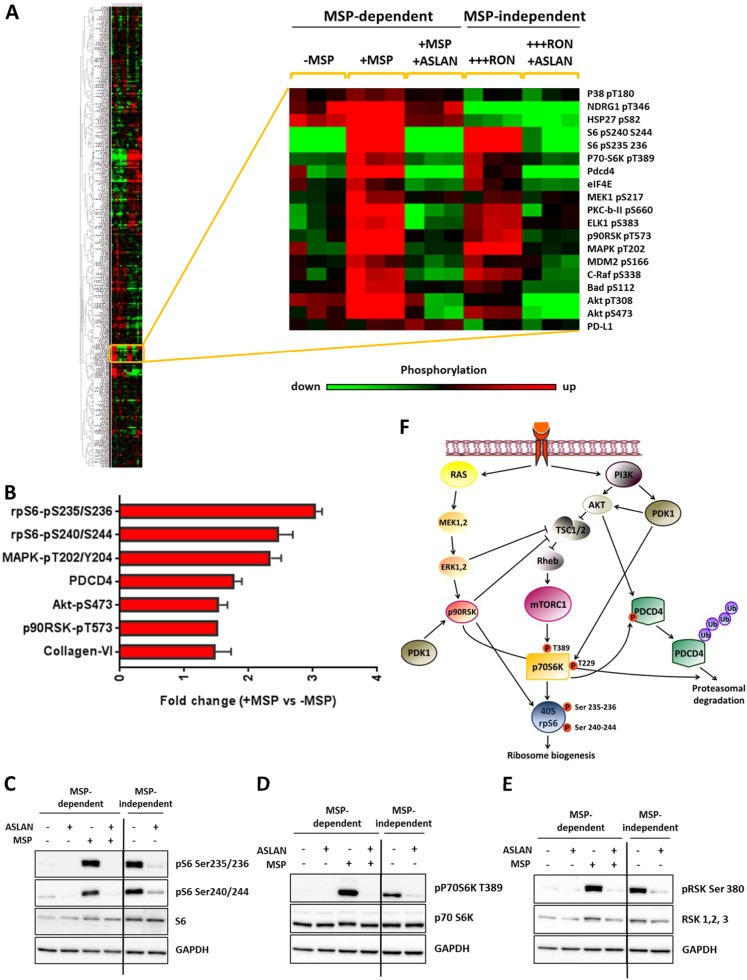
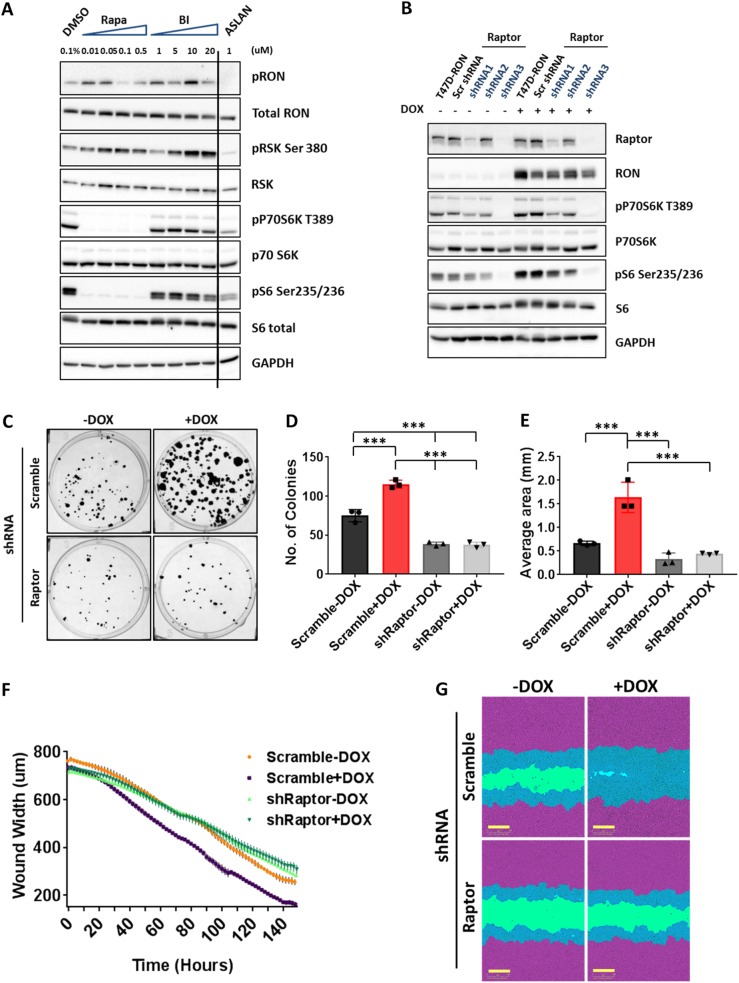
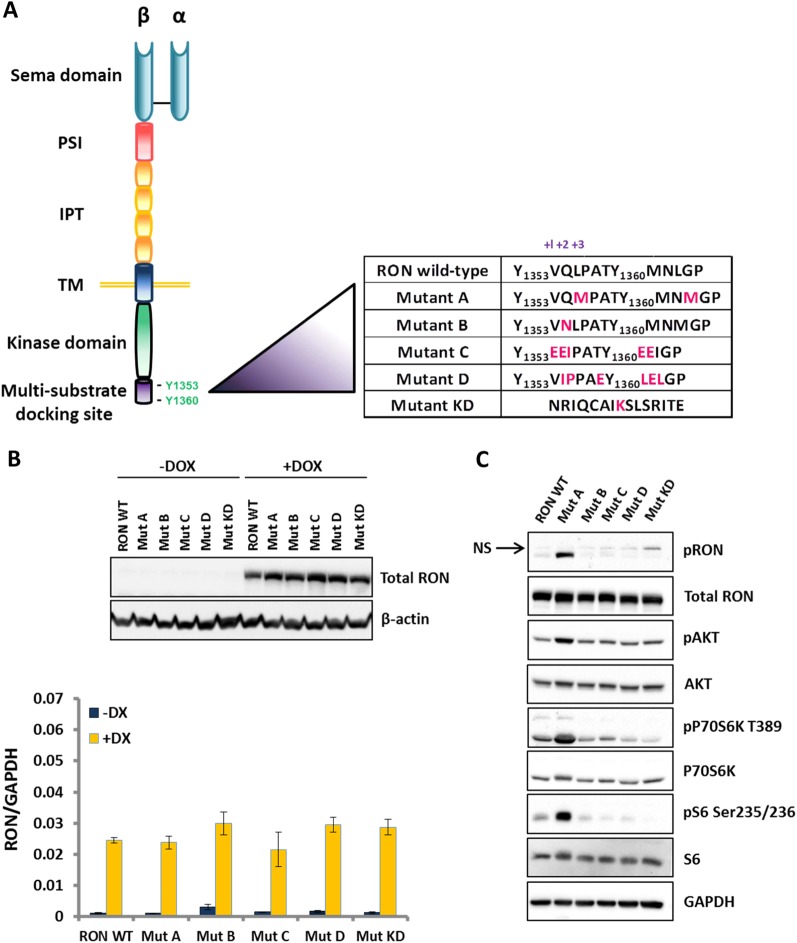
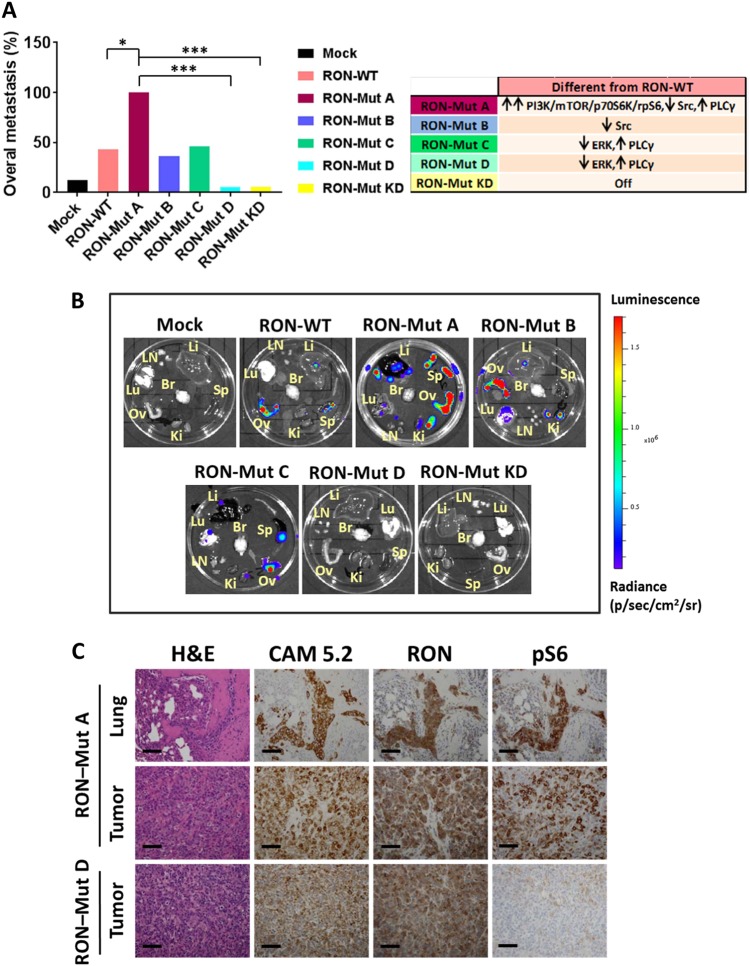
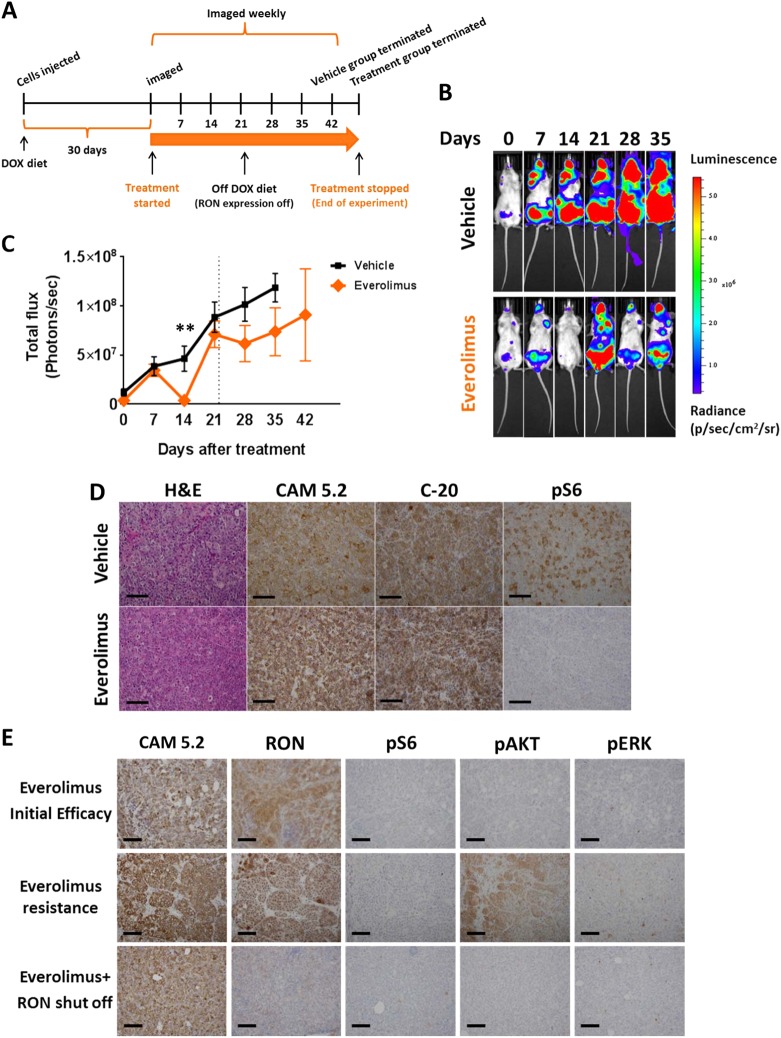
Metastasis is the biggest challenge in treating breast cancer, and it kills >40,000 breast cancer patients annually in the US. Aberrant expression of the RON receptor tyrosine kinase in breast tumors correlates with poor prognosis and has been shown to promote metastasis. However, the molecular mechanisms that govern how RON promotes metastasis, and how to block it, are still largely unknown. We sought to determine critical effectors of RON using a combination of mutational and pharmacologic strategies. High-throughput proteomic analysis of breast cancer cells upon activation of RON showed robust phosphorylation of ribosomal protein S6. Further analysis revealed that RON strongly signals through mTORC1/p70S6K, which is mediated predominantly by the PI3K pathway. A targeted mutation approach to modulate RON signaling validated the importance of PI3K/mTORC1 pathway for spontaneous metastasis in vivo. Finally, inhibition of mTORC1 with an FDA-approved drug, everolimus, resulted in transient shrinkage of established RON-dependent metastases, and combined blockade of mTORC1 and RON delayed progression. These studies have identified a key downstream mediator of RON-dependent metastasis in breast cancer cells and revealed that inhibition of mTORC1, or combined inhibition of mTORC1 and RON, may be effective for treatment of metastatic breast cancers with elevated expression of RON.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
