

Sulfated Non-Saccharide Glycosaminoglycan Mimetics as Novel Drug Discovery Platform for Various Pathologies
- PMID: 30457046
- PMCID: PMC6551317
- DOI: 10.2174/0929867325666181120101147
Sulfated Non-Saccharide Glycosaminoglycan Mimetics as Novel Drug Discovery Platform for Various Pathologies
Abstract
Glycosaminoglycans (GAGs) are very complex, natural anionic polysaccharides. They are polymers of repeating disaccharide units of uronic acid and hexosamine residues. Owing to their template-free, spatiotemporally-controlled, and enzyme-mediated biosyntheses, GAGs possess enormous polydispersity, heterogeneity, and structural diversity which often translate into multiple biological roles. It is well documented that GAGs contribute to physiological and pathological processes by binding to proteins including serine proteases, serpins, chemokines, growth factors, and microbial proteins. Despite advances in the GAG field, the GAG-protein interface remains largely unexploited by drug discovery programs. Thus, Non-Saccharide Glycosaminoglycan Mimetics (NSGMs) have been rationally developed as a novel class of sulfated molecules that modulate GAG-protein interface to promote various biological outcomes of substantial benefit to human health. In this review, we describe the chemical, biochemical, and pharmacological aspects of recently reported NSGMs and highlight their therapeutic potentials as structurally and mechanistically novel anti-coagulants, anti-cancer agents, anti-emphysema agents, and anti-viral agents. We also describe the challenges that complicate their advancement and describe ongoing efforts to overcome these challenges with the aim of advancing the novel platform of NSGMs to clinical use.
Keywords: Glycosaminoglycans; anticancer; anticoagulants; antivirals; non-saccharide glycosaminoglycan mimetics; sulfated molecules..
Copyright© Bentham Science Publishers; For any queries, please email at epub@benthamscience.net.
Conflict of interest statement
CONFLICT OF INTEREST
Authors declare no competing financial conflict of interest.
Figures






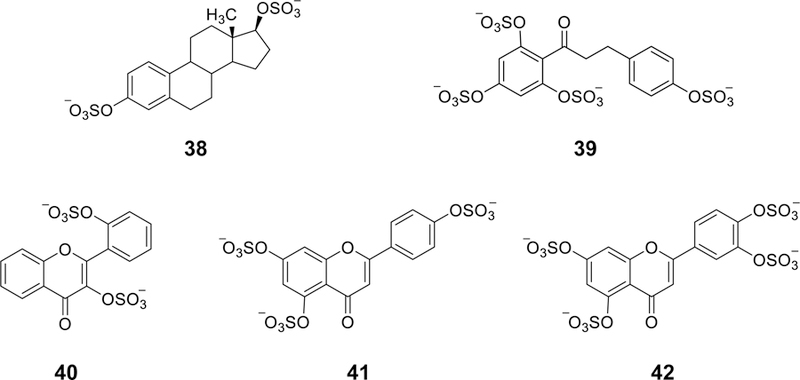
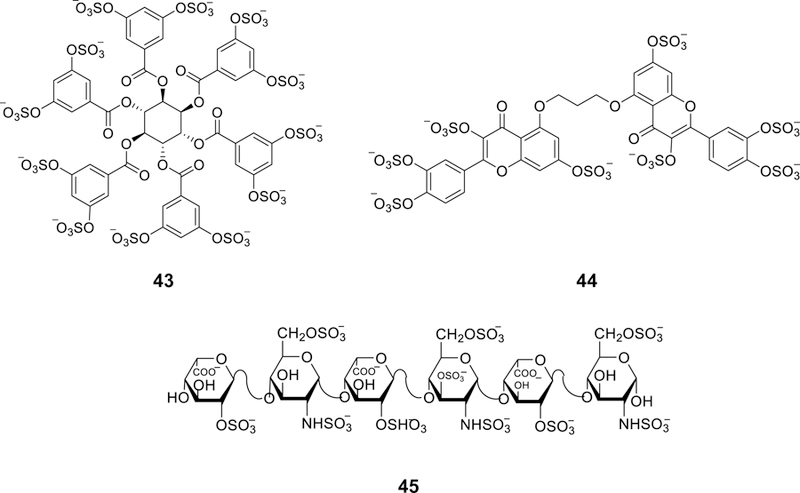

References
-
- Gandhi NS; Mancera RL The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des, 2008, 72, 455–482. - PubMed
-
- Esko JD; Kimata K; Lindahl U Proteoglycans and Sulfated Glycosaminoglycans, In: Essentials of Glycobiology; Varki Ajit, Executive Editor, Cummings Richard D, Esko Jeffrey D, Stanley Pamela, Hart Gerald W, Aebi Markus, Darvill Alan G, Kinoshita Taroh, Packer Nicolle H, Prestegard James H, Schnaar Ronald L, and Seeberger Peter H., Ed.; Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, 2009; pp. 784. - PubMed
-
- Imberty A; Lortat-Jacob H; Pérez S Structural view of glycosaminoglycan-protein interactions. Carbohydr. Res, 2007, 342, 430–439. - PubMed
-
- Sasisekharan R; Venkataraman G Heparin and heparan sulfate: Biosynthesis, structure and function. Curr. Opin. Chem. Biol, 2000, 4, 626–631. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
