Prion Seeds Distribute throughout the Eyes of Sporadic Creutzfeldt-Jakob Disease Patients
- PMID: 30459197
- PMCID: PMC6247090
- DOI: 10.1128/mBio.02095-18
Prion Seeds Distribute throughout the Eyes of Sporadic Creutzfeldt-Jakob Disease Patients
Abstract
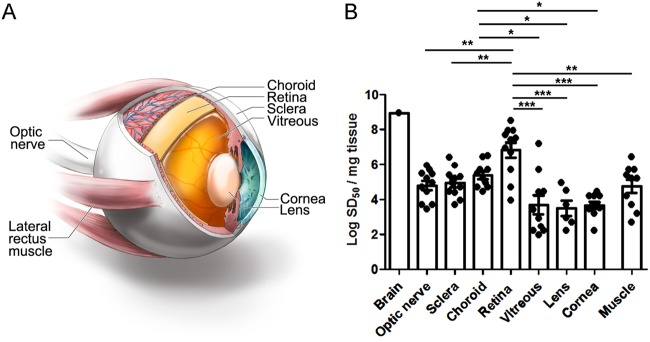
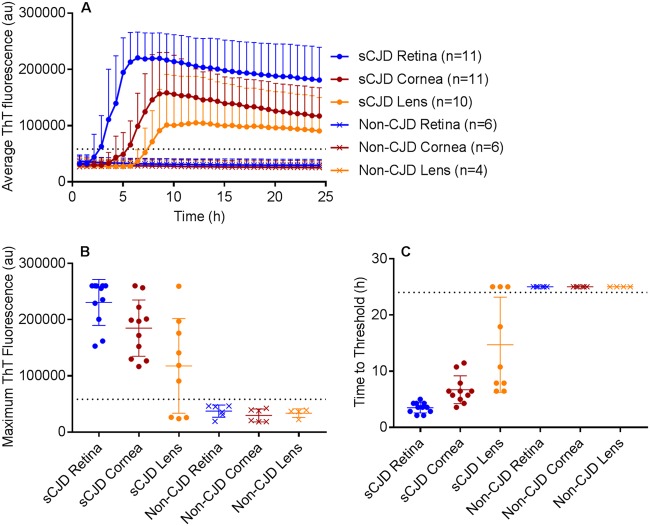
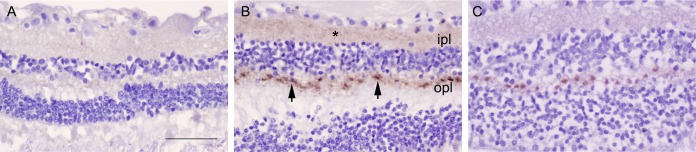
Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common prion disease in humans and has been iatrogenically transmitted through corneal graft transplantation. Approximately 40% of sCJD patients develop visual or oculomotor symptoms and may seek ophthalmological consultation. Here we used the highly sensitive real-time quaking-induced conversion (RT-QuIC) assay to measure postmortem prion seeding activities in cornea, lens, ocular fluid, retina, choroid, sclera, optic nerve, and extraocular muscle in the largest series of sCJD patient eyes studied by any assay to date. We detected prion seeding activity in 100% of sCJD eyes, representing three common sCJD subtypes, with levels varying by up to 4 log-fold among individuals. The retina consistently showed the highest seed levels, which in some cases were only slightly lower than brain. Within the retina, prion deposits were detected by immunohistochemistry (IHC) in the retinal outer plexiform layer in most sCJD cases, and in some eyes the inner plexiform layer, consistent with synaptic prion deposition. Prions were not detected by IHC in any other eye region. With RT-QuIC, prion seed levels generally declined in eye tissues with increased distance from the brain, and yet all corneas had prion seeds detectable. Prion seeds were also present in the optic nerve, extraocular muscle, choroid, lens, vitreous, and sclera. Collectively, these results reveal that sCJD patients accumulate prion seeds throughout the eye, indicating the potential diagnostic utility as well as a possible biohazard.IMPORTANCE Cases of iatrogenic prion disease have been reported from corneal transplants, yet the distribution and levels of prions throughout the eye remain unknown. This study probes the occurrence, level, and distribution of prions in the eyes of patients with sporadic Creutzfeldt-Jakob disease (sCJD). We tested the largest series of prion-infected eyes reported to date using an ultrasensitive technique to establish the prion seed levels in eight regions of the eye. All 11 cases had detectable prion seeds in the eye, and in some cases, the seed levels in the retina approached those in brain. In most cases, prion deposits could also be seen by immunohistochemical staining of retinal tissue; other ocular tissues were negative. Our results have implications for estimating the risk for iatrogenic transmission of sCJD as well as for the development of antemortem diagnostic tests for prion diseases.
Keywords: Creutzfeldt-Jakob disease; RT-QuIC; eye; prion.
Figures