

p66Shc activation promotes increased oxidative phosphorylation and renders CNS cells more vulnerable to amyloid beta toxicity
- PMID: 30459314
- PMCID: PMC6244282
- DOI: 10.1038/s41598-018-35114-y
p66Shc activation promotes increased oxidative phosphorylation and renders CNS cells more vulnerable to amyloid beta toxicity
Abstract
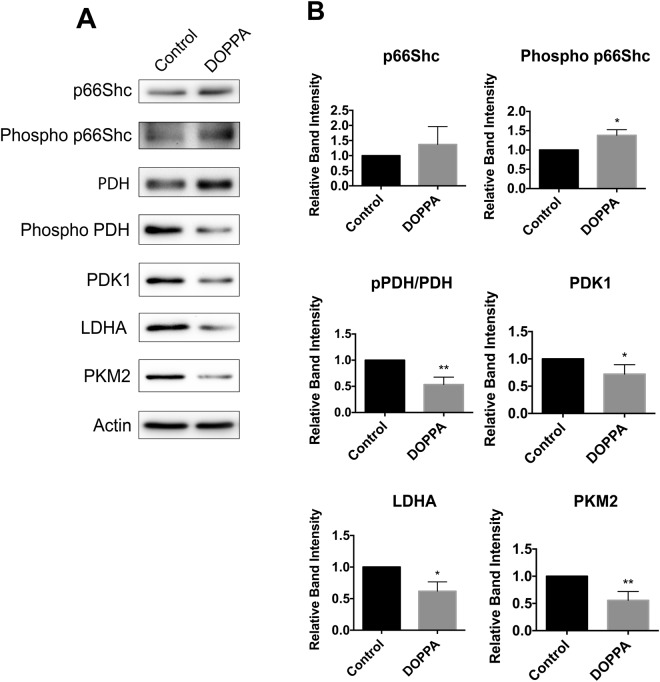
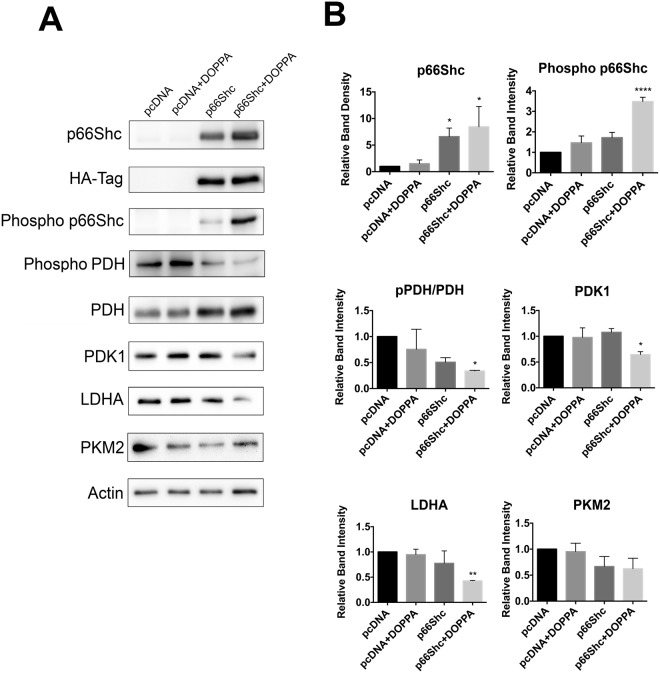
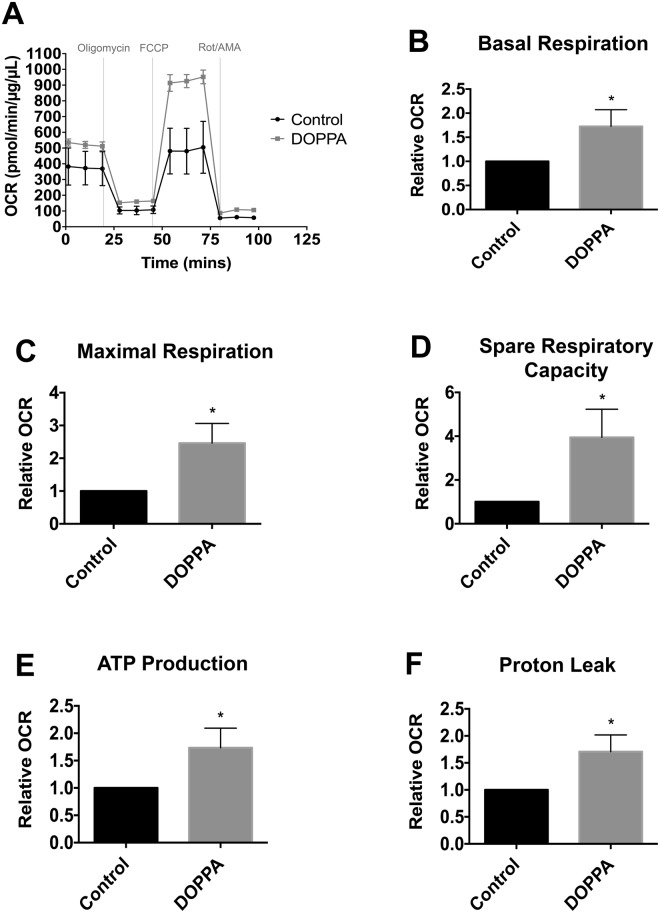
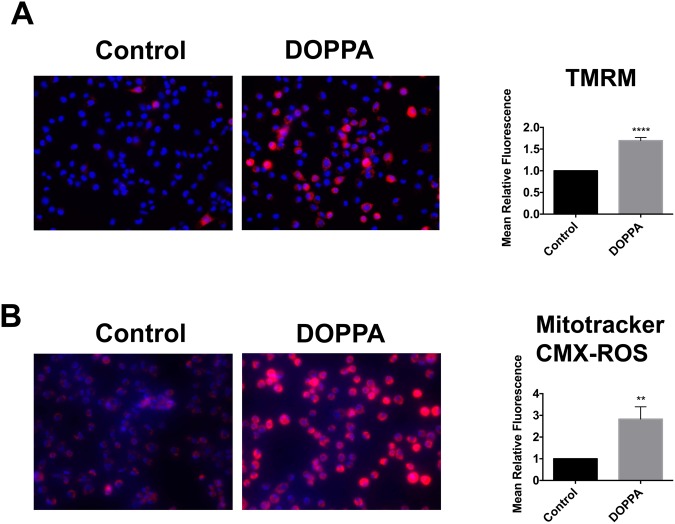
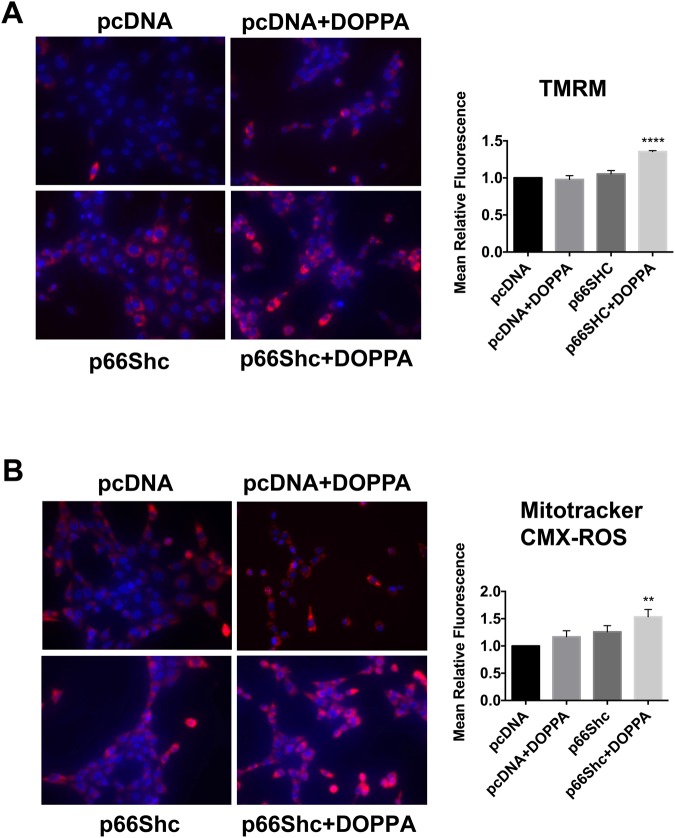
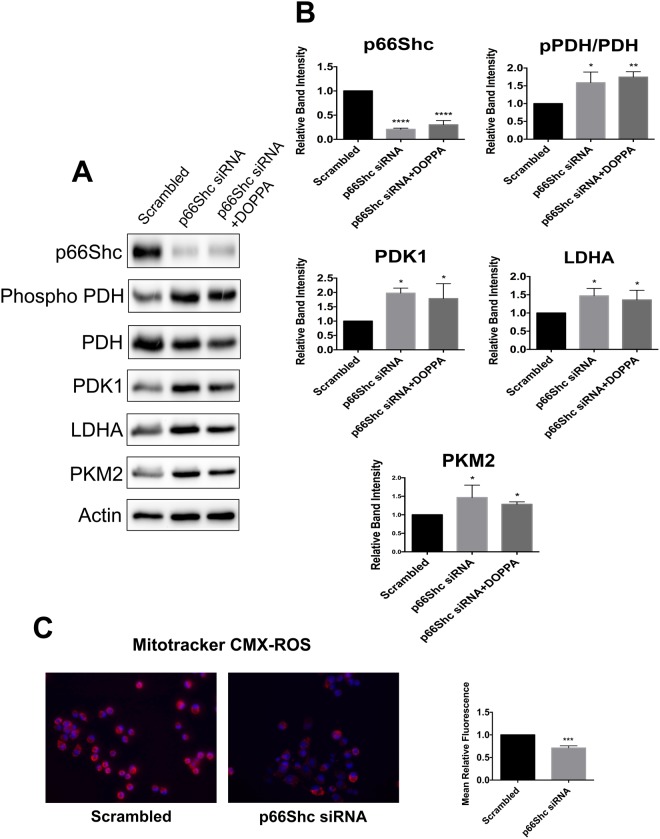
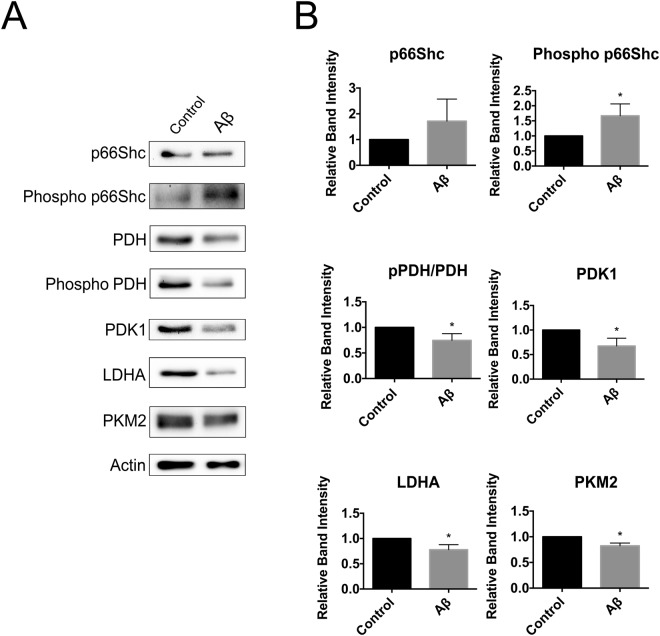
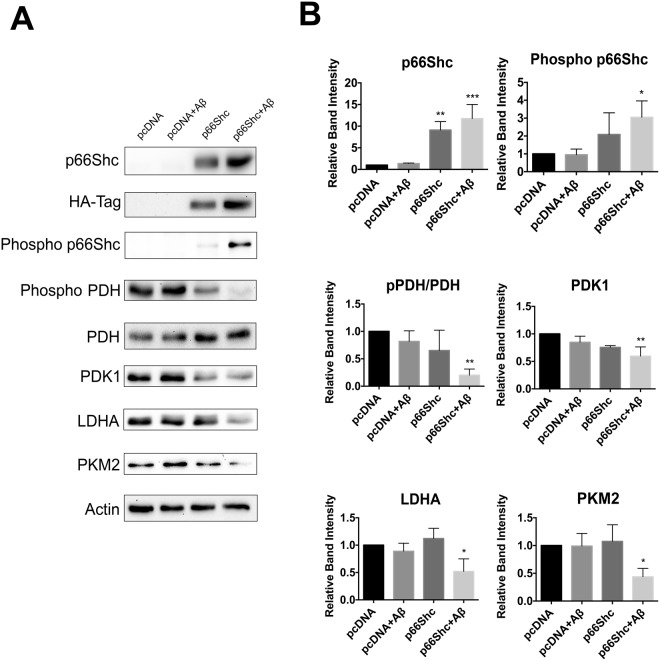
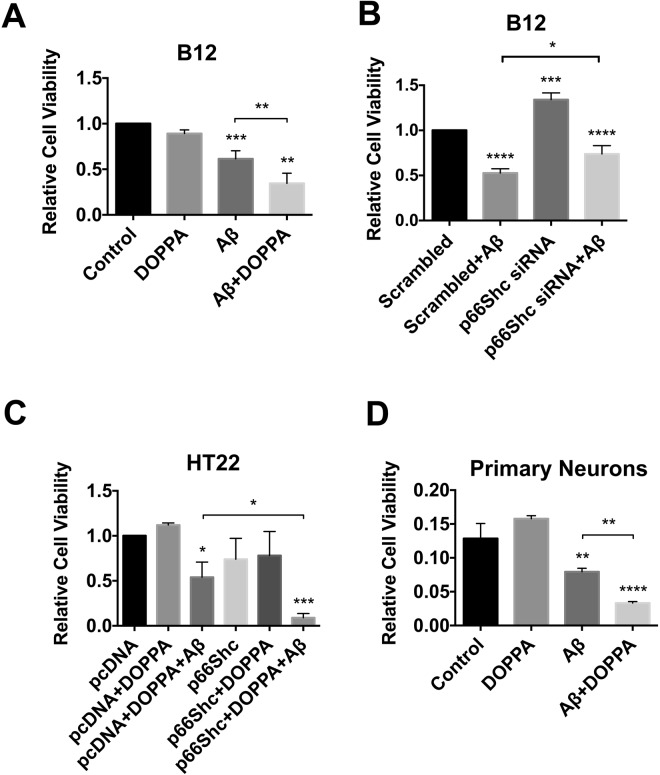
A key pathological feature of Alzheimer's disease (AD) is the accumulation of the neurotoxic amyloid beta (Aβ) peptide within the brains of affected individuals. Previous studies have shown that neuronal cells selected for resistance to Aβ toxicity display a metabolic shift from mitochondrial-dependent oxidative phosphorylation (OXPHOS) to aerobic glycolysis to meet their energy needs. The Src homology/collagen (Shc) adaptor protein p66Shc is a key regulator of mitochondrial function, ROS production and aging. Moreover, increased expression and activation of p66Shc promotes a shift in the cellular metabolic state from aerobic glycolysis to OXPHOS in cancer cells. Here we evaluated the hypothesis that activation of p66Shc in CNS cells promotes both increased OXPHOS and enhanced sensitivity to Aβ toxicity. The effect of altered p66Shc expression on metabolic activity was assessed in rodent HT22 and B12 cell lines of neuronal and glial origin respectively. Overexpression of p66Shc repressed glycolytic enzyme expression and increased both mitochondrial electron transport chain activity and ROS levels in HT22 cells. The opposite effect was observed when endogenous p66Shc expression was knocked down in B12 cells. Moreover, p66Shc activation in both cell lines increased their sensitivity to Aβ toxicity. Our findings indicate that expression and activation of p66Shc renders CNS cells more sensitive to Aβ toxicity by promoting mitochondrial OXPHOS and ROS production while repressing aerobic glycolysis. Thus, p66Shc may represent a potential therapeutically relevant target for the treatment of AD.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity.J Cell Biol. 2005 Apr 25;169(2):331-9. doi: 10.1083/jcb.200410041. Epub 2005 Apr 18. J Cell Biol. 2005. PMID: 15837797 Free PMC article.
-
The oxidoreductase p66Shc acts as tumor suppressor in BRAFV600E-transformed cells.Mol Oncol. 2018 Jun;12(6):869-882. doi: 10.1002/1878-0261.12199. Epub 2018 May 5. Mol Oncol. 2018. PMID: 29624862 Free PMC article.
-
CRIF1 deficiency induces p66shc-mediated oxidative stress and endothelial activation.PLoS One. 2014 Jun 6;9(6):e98670. doi: 10.1371/journal.pone.0098670. eCollection 2014. PLoS One. 2014. PMID: 24906005 Free PMC article.
-
A disease with a sweet tooth: exploring the Warburg effect in Alzheimer's disease.Biogerontology. 2017 Jun;18(3):301-319. doi: 10.1007/s10522-017-9692-x. Epub 2017 Mar 17. Biogerontology. 2017. PMID: 28314935 Review.
-
P66Shc-SIRT1 Regulation of Oxidative Stress Protects Against Cardio-cerebral Vascular Disease.Mol Neurobiol. 2017 Sep;54(7):5277-5285. doi: 10.1007/s12035-016-0073-2. Epub 2016 Aug 30. Mol Neurobiol. 2017. PMID: 27578018 Review.
Cited by
-
Epigenetic mechanisms in cardiovascular complications of diabetes: towards future therapies.Mol Med. 2024 Sep 27;30(1):161. doi: 10.1186/s10020-024-00939-z. Mol Med. 2024. PMID: 39333854 Free PMC article. Review.
-
The p66Shc Redox Protein and the Emerging Complications of Diabetes.Int J Mol Sci. 2023 Dec 20;25(1):108. doi: 10.3390/ijms25010108. Int J Mol Sci. 2023. PMID: 38203279 Free PMC article. Review.
-
P66Shc: A Pleiotropic Regulator of B Cell Trafficking and a Gatekeeper in Chronic Lymphocytic Leukemia.Cancers (Basel). 2020 Apr 19;12(4):1006. doi: 10.3390/cancers12041006. Cancers (Basel). 2020. PMID: 32325830 Free PMC article. Review.
-
Brain aerobic glycolysis and resilience in Alzheimer disease.Proc Natl Acad Sci U S A. 2023 Feb 14;120(7):e2212256120. doi: 10.1073/pnas.2212256120. Epub 2023 Feb 6. Proc Natl Acad Sci U S A. 2023. PMID: 36745794 Free PMC article.
-
p66Shc in Cardiovascular Pathology.Cells. 2022 Jun 6;11(11):1855. doi: 10.3390/cells11111855. Cells. 2022. PMID: 35681549 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
