

A small molecule approach to degrade RAS with EGFR repression is a potential therapy for KRAS mutation-driven colorectal cancer resistance to cetuximab
- PMID: 30459318
- PMCID: PMC6244225
- DOI: 10.1038/s12276-018-0182-2
A small molecule approach to degrade RAS with EGFR repression is a potential therapy for KRAS mutation-driven colorectal cancer resistance to cetuximab
Abstract
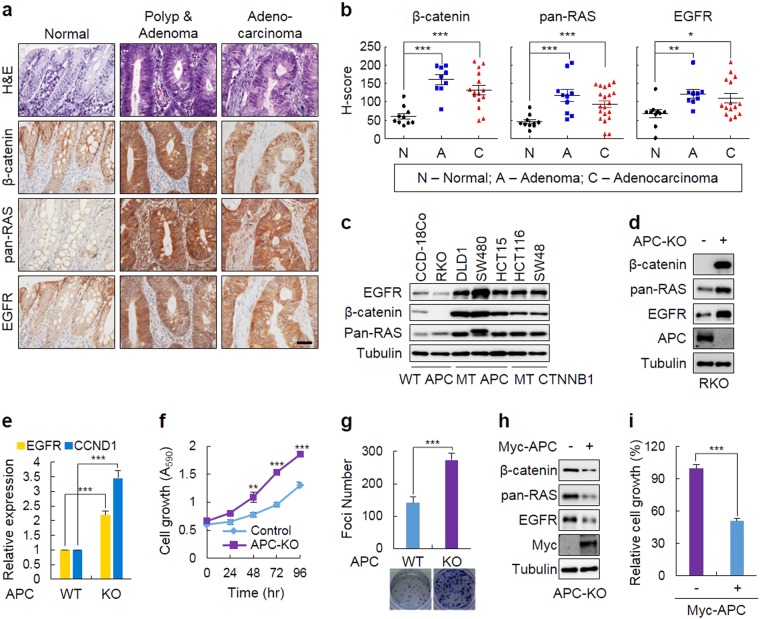
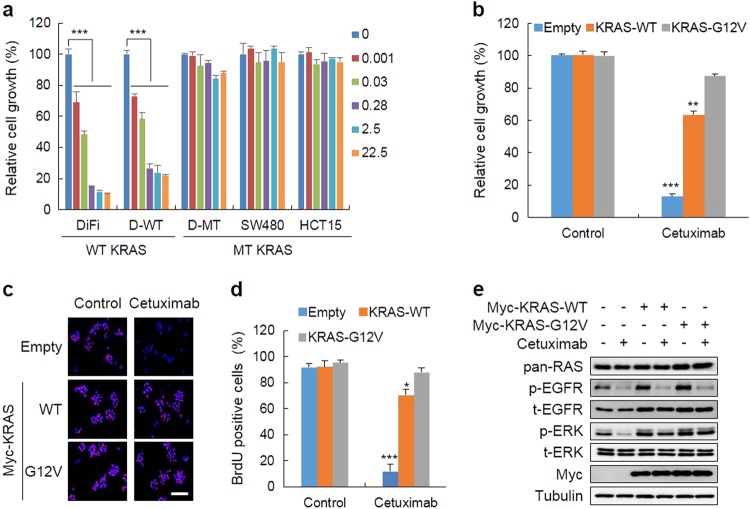
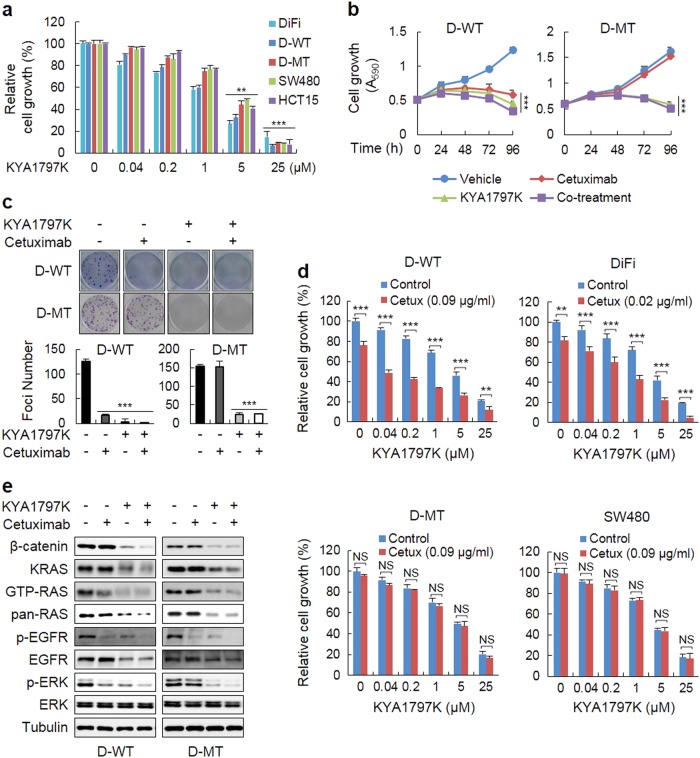
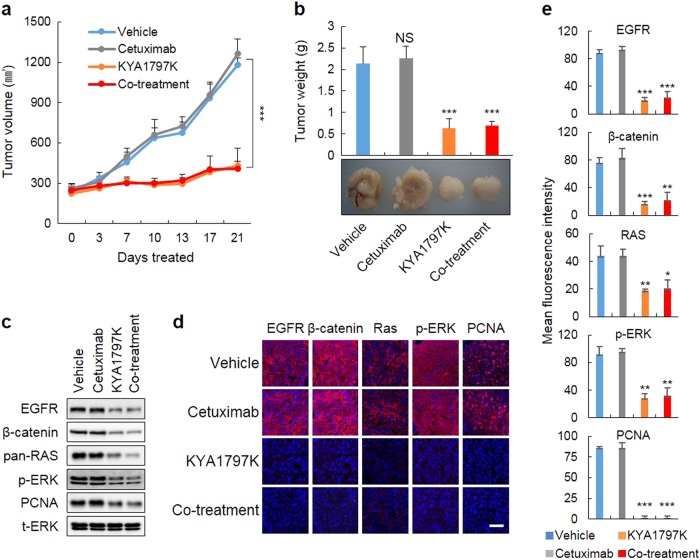
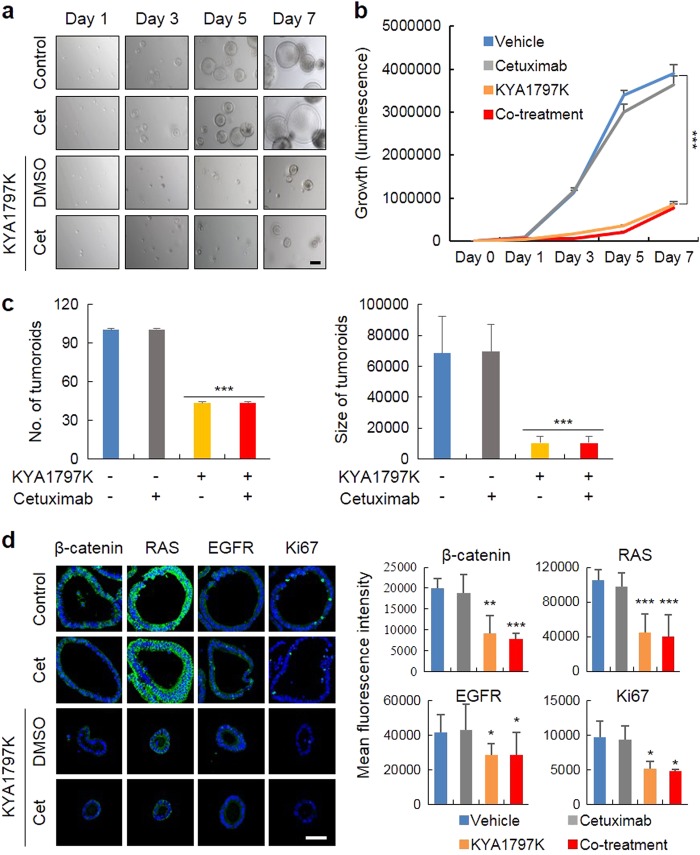
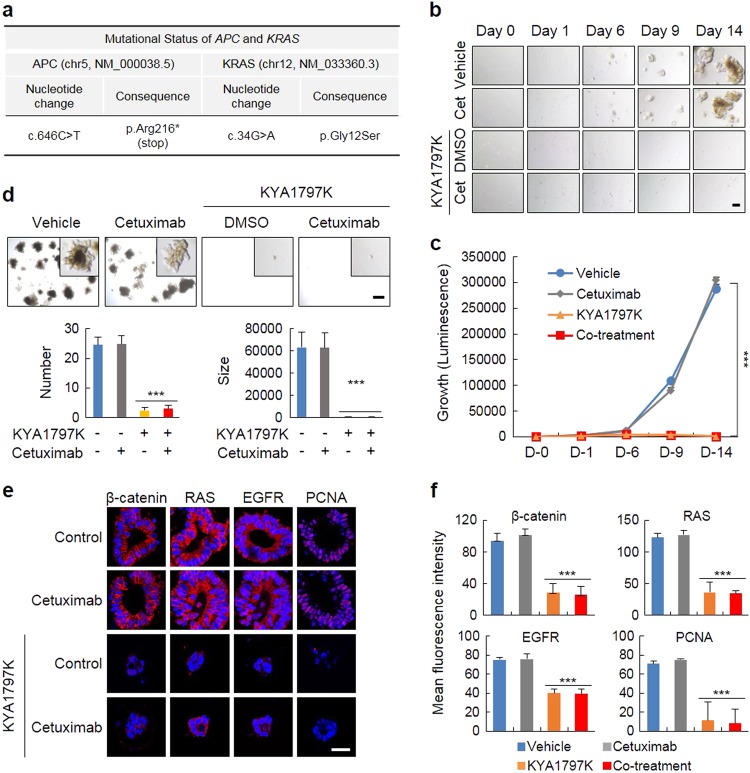
Drugs targeting the epidermal growth factor receptor (EGFR), such as cetuximab and panitumumab, have been prescribed for metastatic colorectal cancer (CRC), but patients harboring KRAS mutations are insensitive to them and do not have an alternative drug to overcome the problem. The levels of β-catenin, EGFR, and RAS, especially mutant KRAS, are increased in CRC patient tissues due to mutations of adenomatous polyposis coli (APC), which occur in 90% of human CRCs. The increases in these proteins by APC loss synergistically promote tumorigenesis. Therefore, we tested KYA1797K, a recently identified small molecule that degrades both β-catenin and Ras via GSK3β activation, and its capability to suppress the cetuximab resistance of KRAS-mutated CRC cells. KYA1797K suppressed the growth of tumor xenografts induced by CRC cells as well as tumor organoids derived from CRC patients having both APC and KRAS mutations. Lowering the levels of both β-catenin and RAS as well as EGFR via targeting the Wnt/β-catenin pathway is a therapeutic strategy for controlling CRC and other types of cancer with aberrantly activated the Wnt/β-catenin and EGFR-RAS pathways, including those with resistance to EGFR-targeting drugs attributed to KRAS mutations.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
