

A ten year review of the sickle cell program in Muhimbili National Hospital, Tanzania
- PMID: 30459954
- PMCID: PMC6236876
- DOI: 10.1186/s12878-018-0125-0
A ten year review of the sickle cell program in Muhimbili National Hospital, Tanzania
Abstract
Background: Africa has the highest burden of Sickle cell disease (SCD) but there are few large, systematic studies providing reliable descriptions of the disease spectrum. Tanzania, with 11,000 SCD births annually, established the Muhimbili Sickle Cell program aiming to improve understanding of SCD in Africa. We report the profile of SCD seen in the first 10 years at Muhimbili National Hospital (MNH).
Methods: Individuals seen at MNH known or suspected to have SCD were enrolled at clinic and laboratory testing for SCD, haematological and biochemical analyses done. Ethnicity was self-reported. Clinical and laboratory features of SCD were documented. Comparison was made with non-SCD population as well as within 3 different age groups (< 5, 5-17 and ≥ 18 years) within the SCD population.
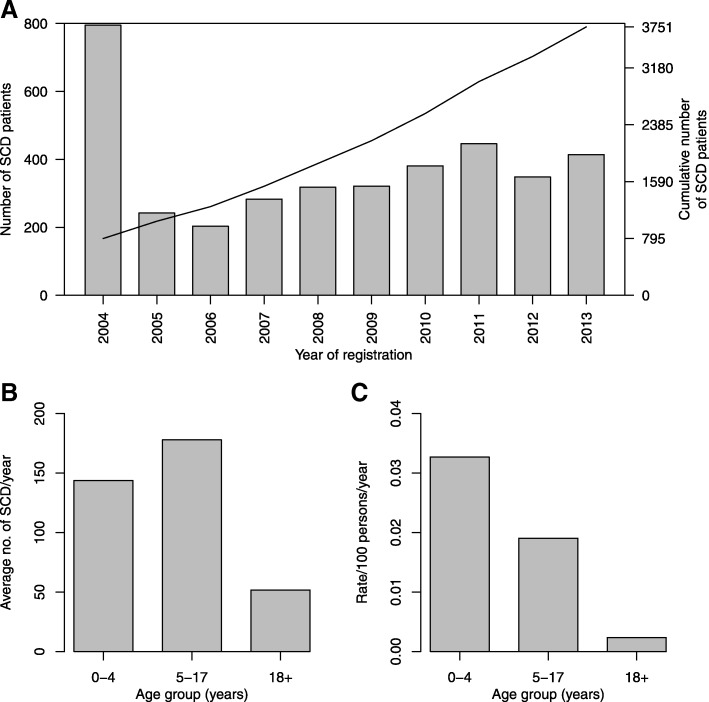
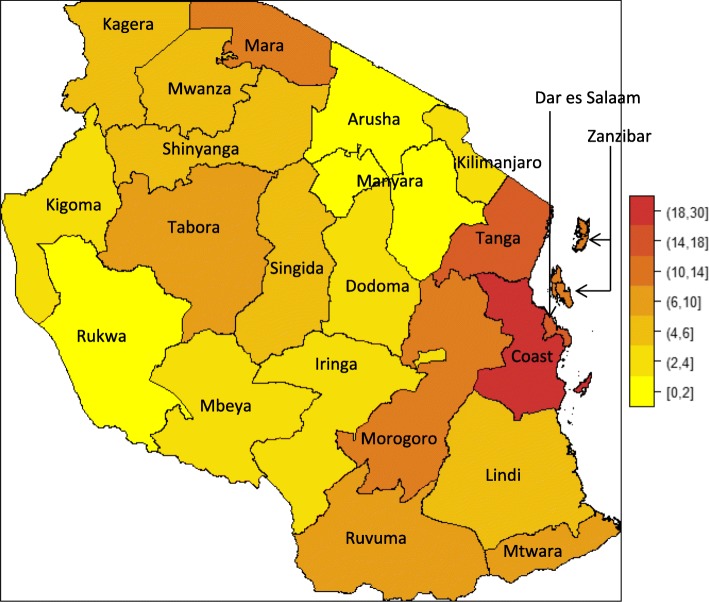
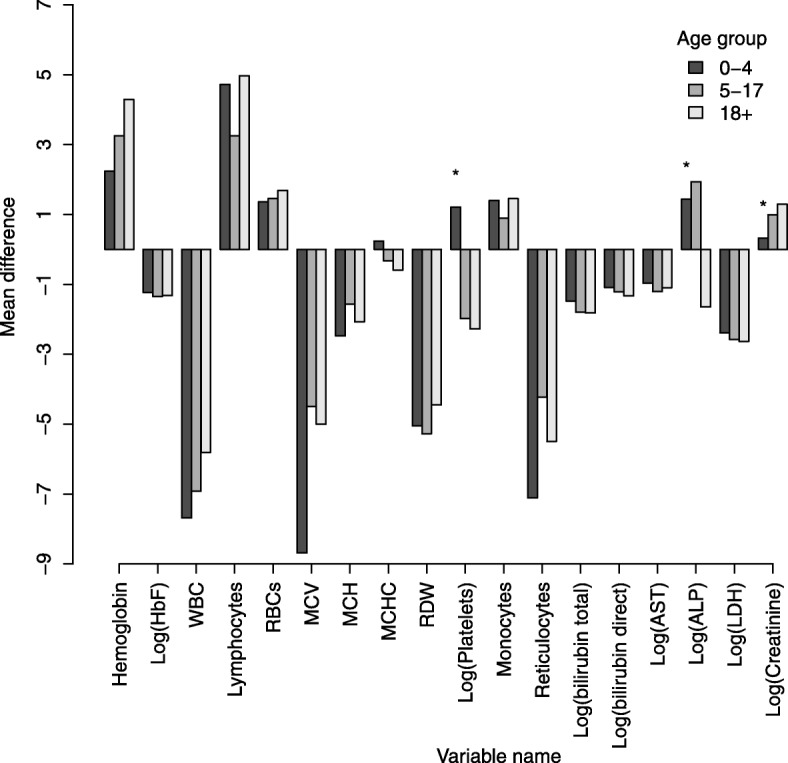
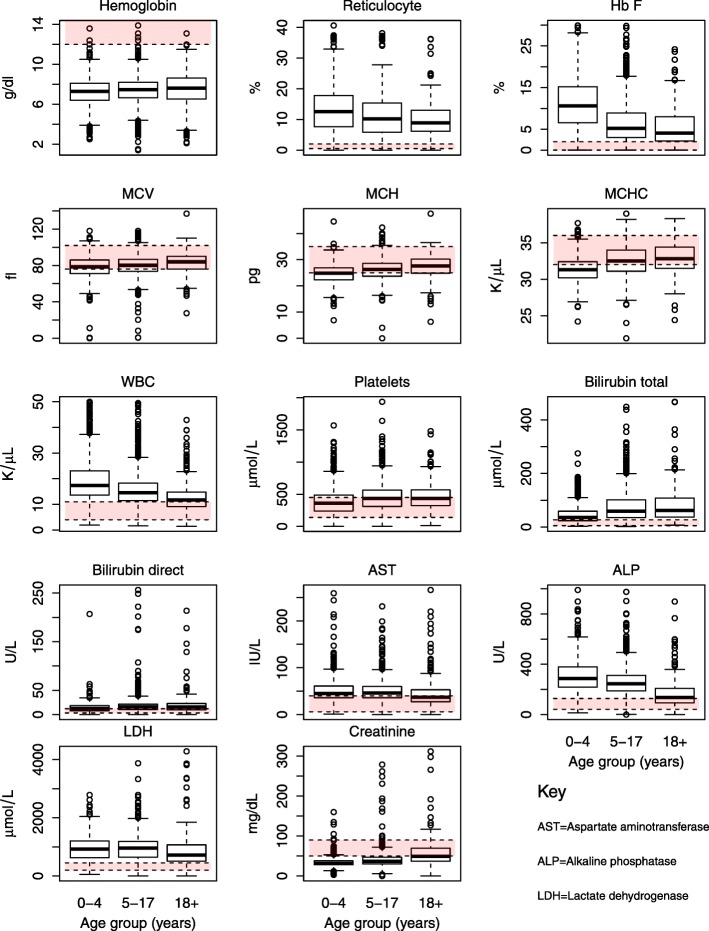
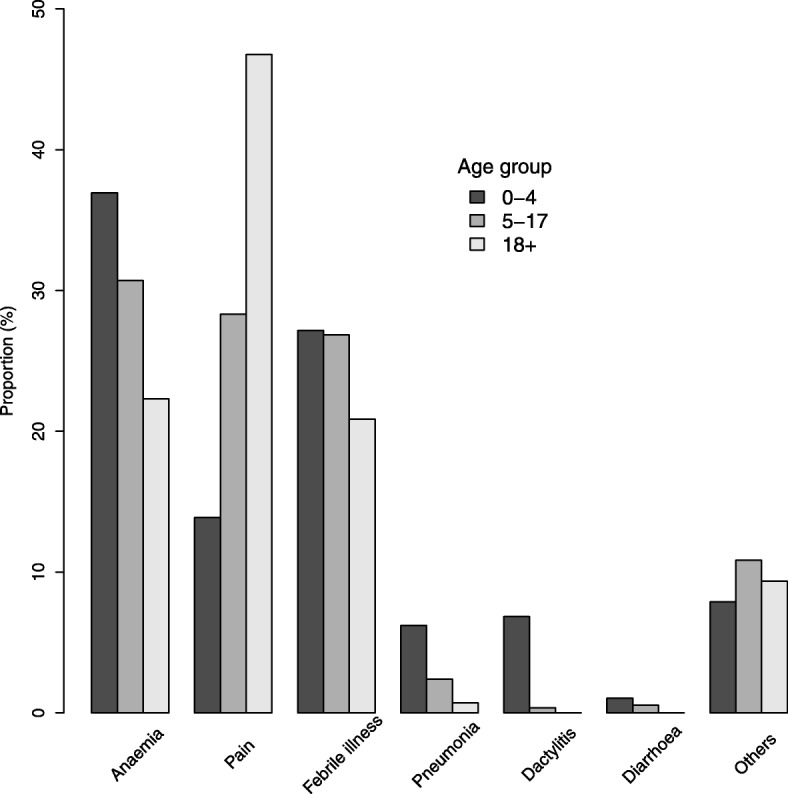
Results: From 2004 to 2013, 6397 individuals, 3751 (58.6%) SCD patients, were enrolled, the majority (47.4%) in age group 5-17 years. There was variation in the geographical distribution of SCD. Individuals with SCD compared to non-SCD, had significantly lower blood pressure and peripheral oxygen saturation (SpO2). SCD patients had higher prevalence of severe anemia, jaundice and desaturation (SpO2 < 95%) as well as higher levels of reticulocytes, white blood cells, platelets and fetal hemoglobin. The main causes of hospitalization for SCD within a 12-month period preceding enrolment were pain (adults), and fever and severe anemia (children). When clinical and laboratory features were compared in SCD within 3 age groups, there was a progressive decrease in the prevalence of splenic enlargement and an increase in prevalence of jaundice. Furthermore, there were significant differences with monotonic trends across age groups in SpO2, hematological and biochemical parameters.
Conclusion: This report confirms that the wide spectrum of clinical expression of SCD observed elsewhere is also present in Tanzania, with non-uniform geographical distribution across the country. Age-specific analysis is consistent with different disease-patterns across the lifespan.
Keywords: Africa; Sickle cell anemia; Tanzania.
Conflict of interest statement
Approval was by MUHAS ethical committee (MU/RP/AEC/VOL XI/33) and permission given by MNH. Written informed consent was obtained from all participants. For individuals 18 years or above, while for minors, a parent/guardian consented and signed the consent on behalf of the patient; adolescents provided assent.Not applicableThe authors declare that they have no competing interests.Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Piel Frédéric B, Patil Anand P, Howes Rosalind E, Nyangiri Oscar A, Gething Peter W, Dewi Mewahyu, Temperley William H, Williams Thomas N, Weatherall David J, Hay Simon I. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. The Lancet. 2013;381(9861):142–151. doi: 10.1016/S0140-6736(12)61229-X. - DOI - PMC - PubMed
-
- Weatherall D, Akinyanju O, Fucharoen S, Olivieri N, Musgrove P. Inherited disorders of hemoglobin. 2006. - PubMed
-
- Gaston M, Rosse WF. The cooperative study of sickle cell disease: review of study design and objectives. Am J Pediatr Hematol Oncol. 1982;4:197–201. - PubMed
-
- Telfer P., Coen P., Chakravorty S., Wilkey O., Evans J., Newell H., Smalling B., Amos R., Stephens A., Rogers D., Kirkham F. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92(7):905–912. doi: 10.3324/haematol.10937. - DOI - PubMed
-
- Gill FM, Sleeper LA, Weiner SJ, Brown AK, Bellevue R, Grover R, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood. 1995;86:776–83. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
