

Exploring glia to better understand Alzheimer's disease
- PMID: 30460100
- PMCID: PMC6138241
- DOI: 10.1080/19768354.2018.1508498
Exploring glia to better understand Alzheimer's disease
Abstract
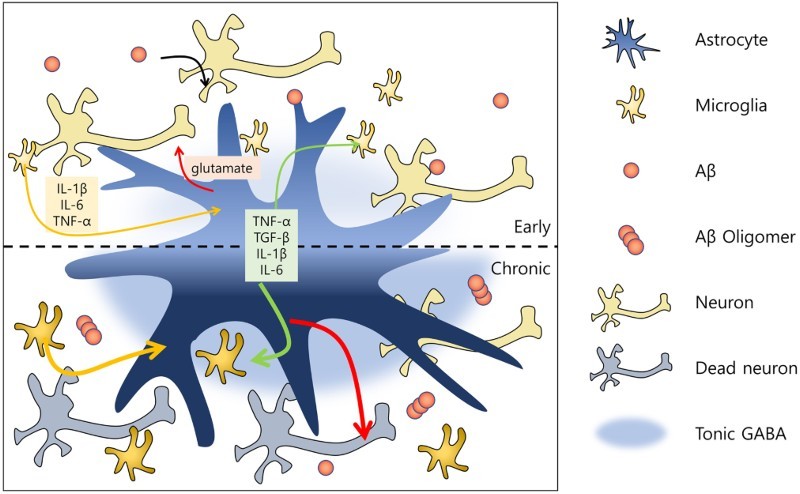
The amyloid-β (Aβ) hypothesis has been the leading explanation for the pathogenesis of Alzheimer's disease (AD). The most common traits of AD are cognitive impairments and memory loss, which are associated with the accumulation of Aβ. Aβ aggregates activate glial cells, which in turn remove Aβ. Because microglia act as immune cells in the brain, most glia-related studies of AD have focused primarily on this cell type. However, astrocytes, another type of glial cell, also participate in the brain immune system, synaptic formation, brain homeostasis, and various other brain functions. Accordingly, many studies on the underlying mechanisms of AD have investigated not only neurons but also glial cells. Although these studies suggest that microglia and astrocytes are effective targets for AD therapeutics, other recent studies have raised questions regarding whether microglial cells and/or astrocytes serve a neuroprotective or neurotoxic function in AD. To gain a better understanding of the mechanisms of AD and identify novel targets for AD treatment, in this review, we consider the role of both microglia and astrocytes in AD.
Keywords: Alzheimer’s disease; astrocytes; microglia; neuroglia.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources