

scFTD-seq: freeze-thaw lysis based, portable approach toward highly distributed single-cell 3' mRNA profiling
- PMID: 30462277
- PMCID: PMC6379653
- DOI: 10.1093/nar/gky1173
scFTD-seq: freeze-thaw lysis based, portable approach toward highly distributed single-cell 3' mRNA profiling
Abstract
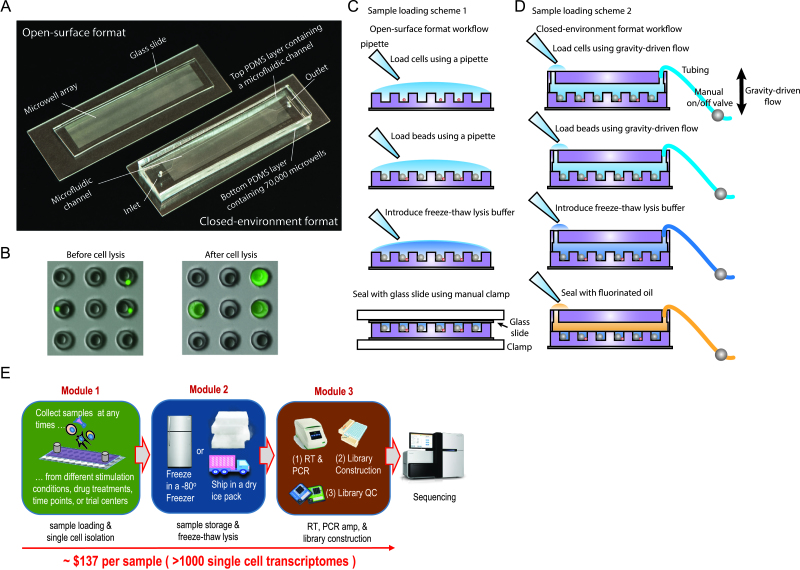
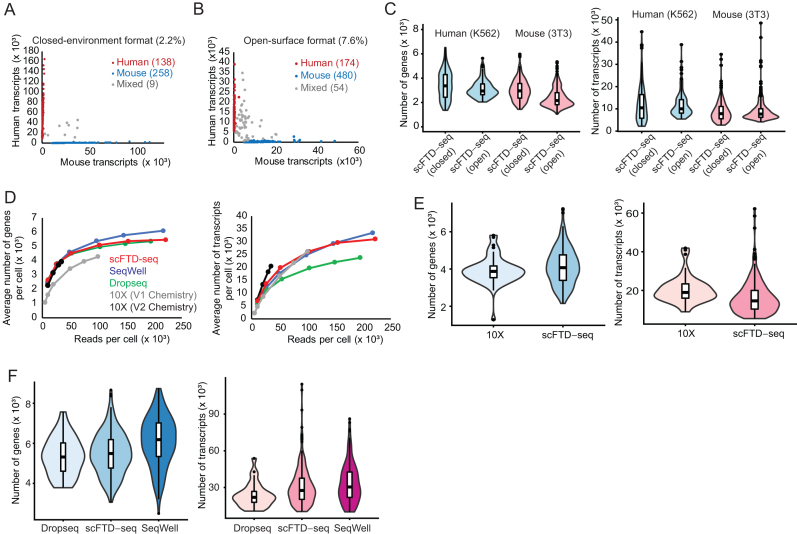
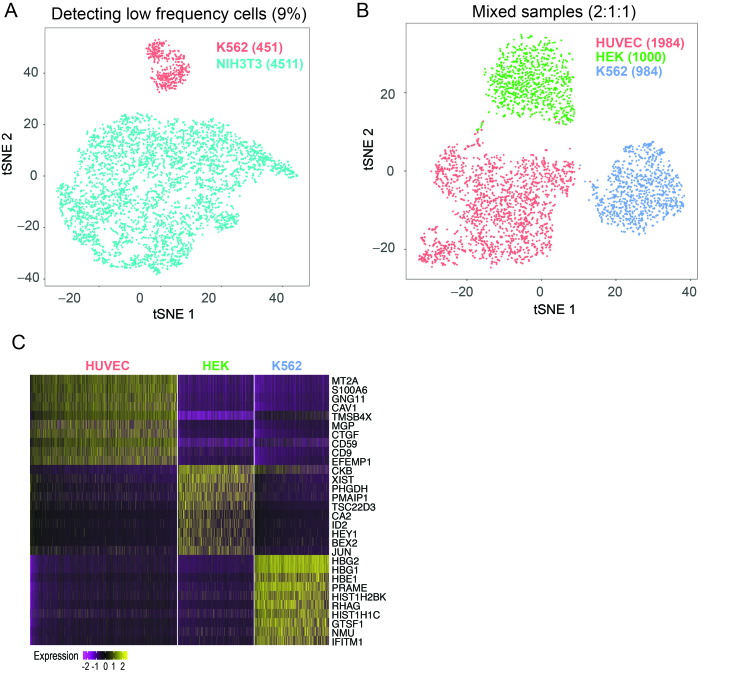
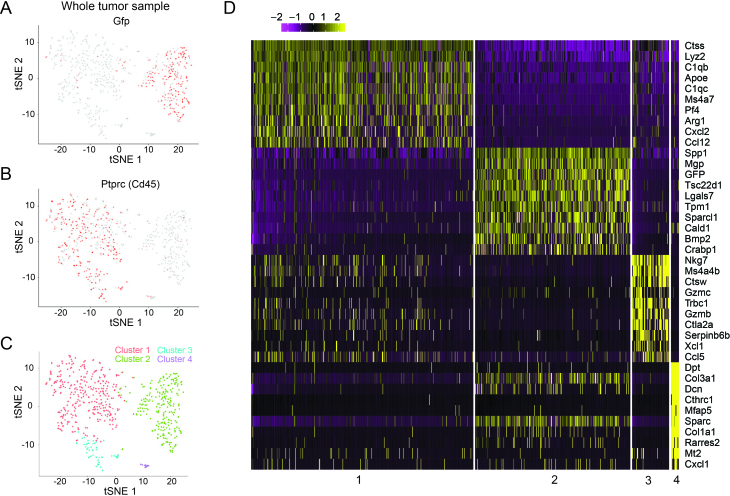
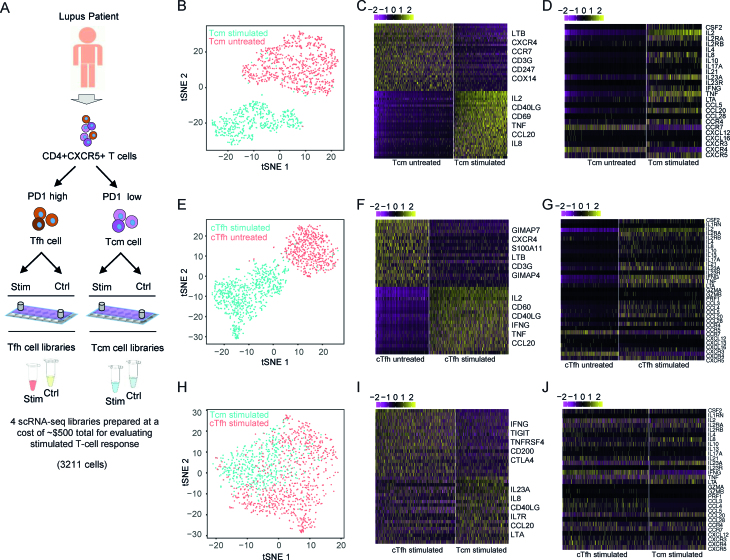
Cellular barcoding of 3' mRNAs enabled massively parallel profiling of single-cell gene expression and has been implemented in droplet and microwell based platforms. The latter further adds the value for compatibility with low input samples, optical imaging, scalability, and portability. However, cell lysis in microwells remains challenging despite the recently developed sophisticated solutions. Here, we present scFTD-seq, a microchip platform for performing single-cell freeze-thaw lysis directly toward 3' mRNA sequencing. It offers format flexibility with a simplified, widely adoptable workflow that reduces the number of preparation steps and hands-on time, with the quality of data and cost per sample matching that of the state-of-the-art scRNA-seq platforms. Freeze-thaw, known as an unfavorable lysis method resulting in possible RNA fragmentation, turns out to be fully compatible with 3' scRNA-seq. We applied it to the profiling of circulating follicular helper T cells implicated in systemic lupus erythematosus pathogenesis. Our results delineate the heterogeneity in the transcriptional programs and effector functions of these rare pathogenic T cells. As scFTD-seq decouples on-chip cell isolation and library preparation, we envision it to allow sampling at the distributed sites including point-of-care settings and downstream processing at centralized facilities, which should enable wide-spread adoption beyond academic laboratories.
© The Author(s) 2018. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
STRT-seq-2i: dual-index 5' single cell and nucleus RNA-seq on an addressable microwell array.Sci Rep. 2017 Nov 27;7(1):16327. doi: 10.1038/s41598-017-16546-4. Sci Rep. 2017. PMID: 29180631 Free PMC article.
-
Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput.Nat Methods. 2017 Apr;14(4):395-398. doi: 10.1038/nmeth.4179. Epub 2017 Feb 13. Nat Methods. 2017. PMID: 28192419 Free PMC article.
-
Seq-Well: A Sample-Efficient, Portable Picowell Platform for Massively Parallel Single-Cell RNA Sequencing.Methods Mol Biol. 2019;1979:111-132. doi: 10.1007/978-1-4939-9240-9_8. Methods Mol Biol. 2019. PMID: 31028635 Free PMC article.
-
Current and Future Methods for mRNA Analysis: A Drive Toward Single Molecule Sequencing.Methods Mol Biol. 2018;1783:209-241. doi: 10.1007/978-1-4939-7834-2_11. Methods Mol Biol. 2018. PMID: 29767365 Review.
-
Approaches for single-cell RNA sequencing across tissues and cell types.Transcription. 2023 Jun-Oct;14(3-5):127-145. doi: 10.1080/21541264.2023.2200721. Epub 2023 Apr 16. Transcription. 2023. PMID: 37062951 Free PMC article. Review.
Cited by
-
Single-cell RNA sequencing reveals changes in glioma-associated macrophage polarization and cellular states of malignant gliomas with high AQP4 expression.Cancer Gene Ther. 2023 May;30(5):716-726. doi: 10.1038/s41417-022-00582-y. Epub 2023 Jan 4. Cancer Gene Ther. 2023. PMID: 36599974 Free PMC article.
-
Navoximod modulates local HSV-1 replication to reshape tumor immune microenvironment for enhanced immunotherapy via an injectable hydrogel.Commun Biol. 2023 Jun 9;6(1):621. doi: 10.1038/s42003-023-04983-z. Commun Biol. 2023. PMID: 37296221 Free PMC article.
-
Identification of novel protein biomarkers from the blood and urine for the early diagnosis of bladder cancer via proximity extension analysis.J Transl Med. 2024 Mar 26;22(1):314. doi: 10.1186/s12967-024-04951-z. J Transl Med. 2024. PMID: 38532419 Free PMC article.
-
Cryoablation-induced modulation of Treg cells and the TGF-β pathway in lung adenocarcinoma: implications for increased antitumor immunity.BMC Med. 2025 Feb 14;23(1):89. doi: 10.1186/s12916-025-03926-1. BMC Med. 2025. PMID: 39948553 Free PMC article.
-
Single-Cell Transcriptome Comparison of Bladder Cancer Reveals Its Ecosystem.Front Oncol. 2022 Feb 21;12:818147. doi: 10.3389/fonc.2022.818147. eCollection 2022. Front Oncol. 2022. PMID: 35265520 Free PMC article.
References
-
- Eberwine J., Sul J.-Y., Bartfai T., Kim J.. The promise of single-cell sequencing. Nat. Methods. 2014; 11:25–27. - PubMed
-
- Björklund Å.K., Forkel M., Picelli S., Konya V., Theorell J., Friberg D., Sandberg R., Mjösberg J.. The heterogeneity of human CD127+ innate lymphoid cells revealed by single-cell RNA sequencing. Nat. Immunol. 2016; 17:451–460. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
