

STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury
- PMID: 30463579
- PMCID: PMC6247615
- DOI: 10.1186/s12974-018-1354-7
STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury
Abstract
Background: Traumatic brain injury (TBI) represents a major cause of disability and death worldwide with sustained neuroinflammation and autophagy dysfunction contributing to the cellular damage. Stimulator of interferon genes (STING)-induced type-I interferon (IFN) signalling is known to be essential in mounting the innate immune response against infections and cell injury in the periphery, but its role in the CNS remains unclear. We previously identified the type-I IFN pathway as a key mediator of neuroinflammation and neuronal cell death in TBI. However, the modulation of the type-I IFN and neuroinflammatory responses by STING and its contribution to autophagy and neuronal cell death after TBI has not been explored.
Methods: C57BL/6J wild-type (WT) and STING-/- mice (8-10-week-old males) were subjected to controlled cortical impact (CCI) surgery and brains analysed by QPCR, Western blot and immunohistochemical analyses at 2 h or 24 h. STING expression was also analysed by QPCR in post-mortem human brain samples.
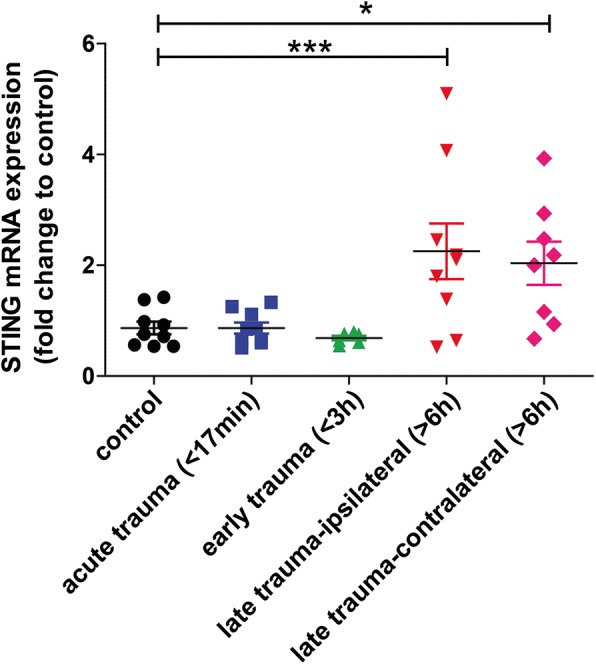
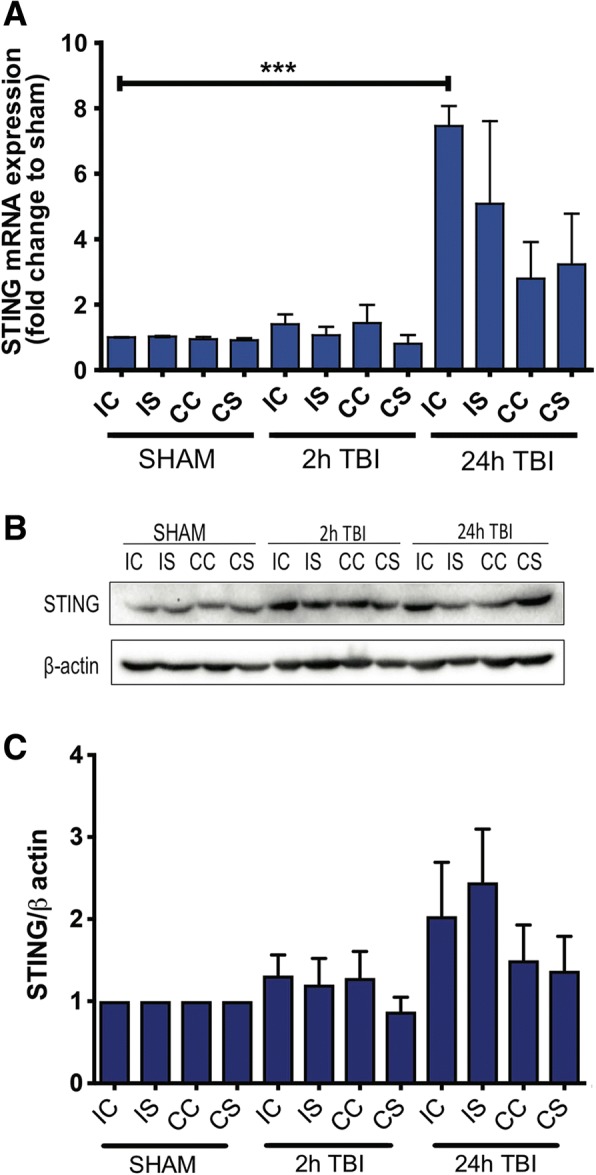
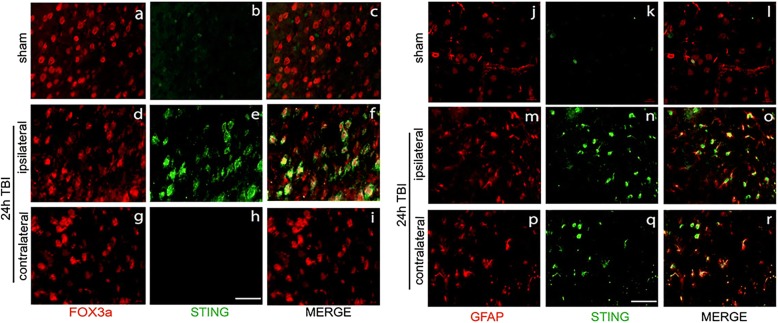
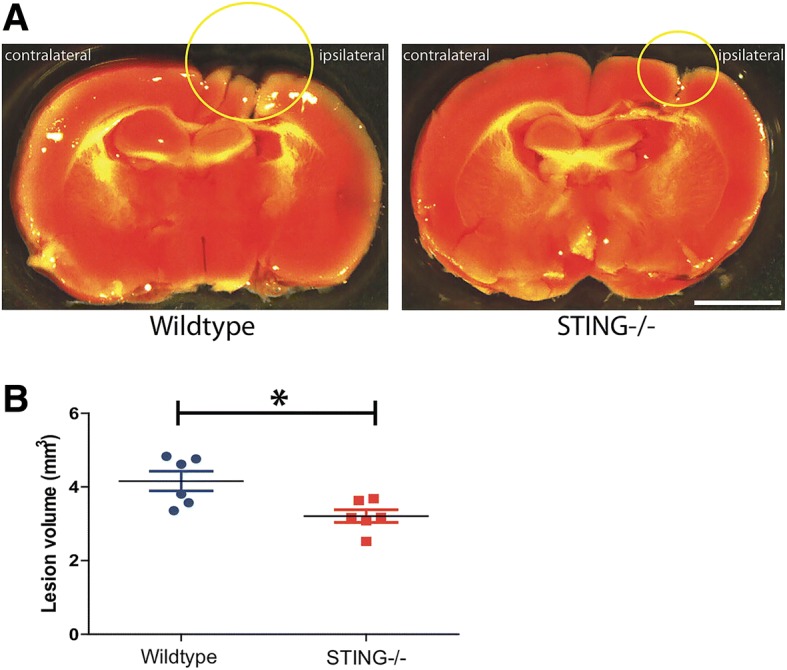
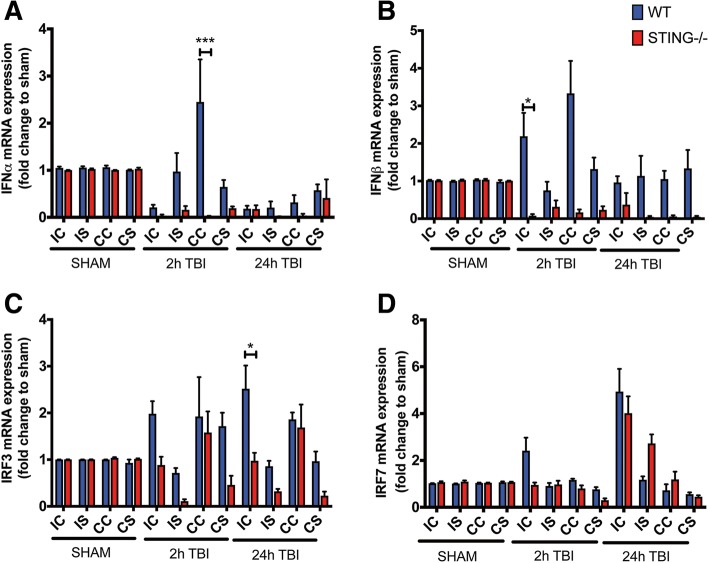
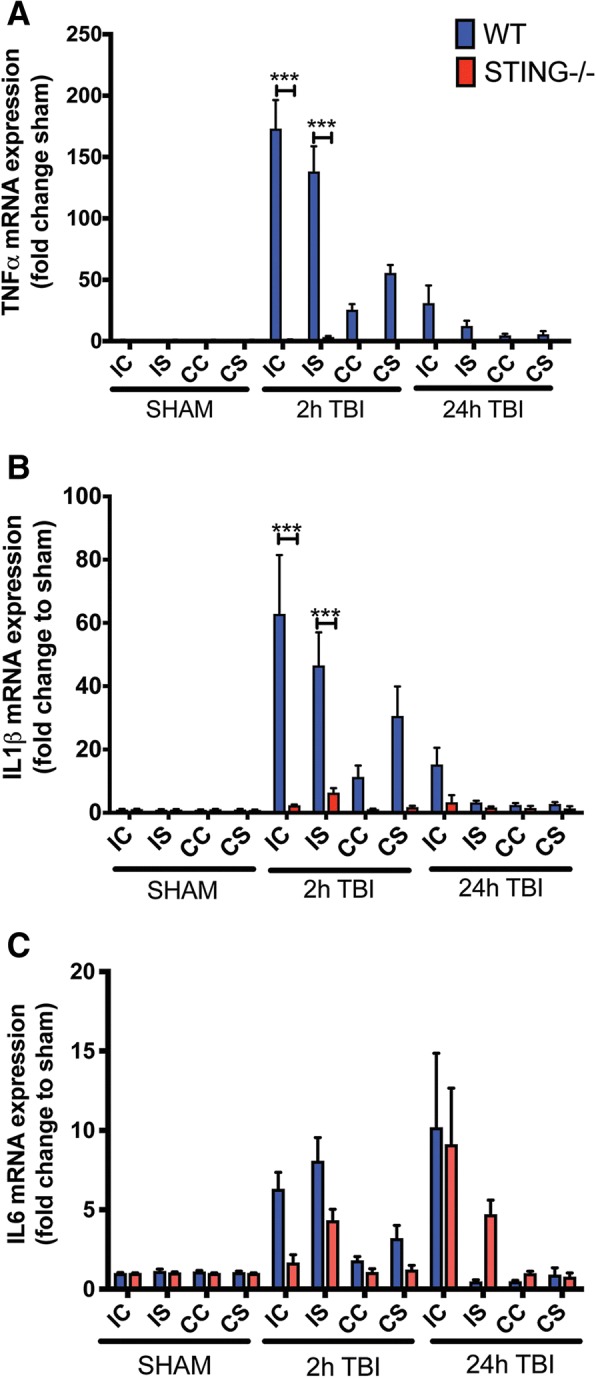
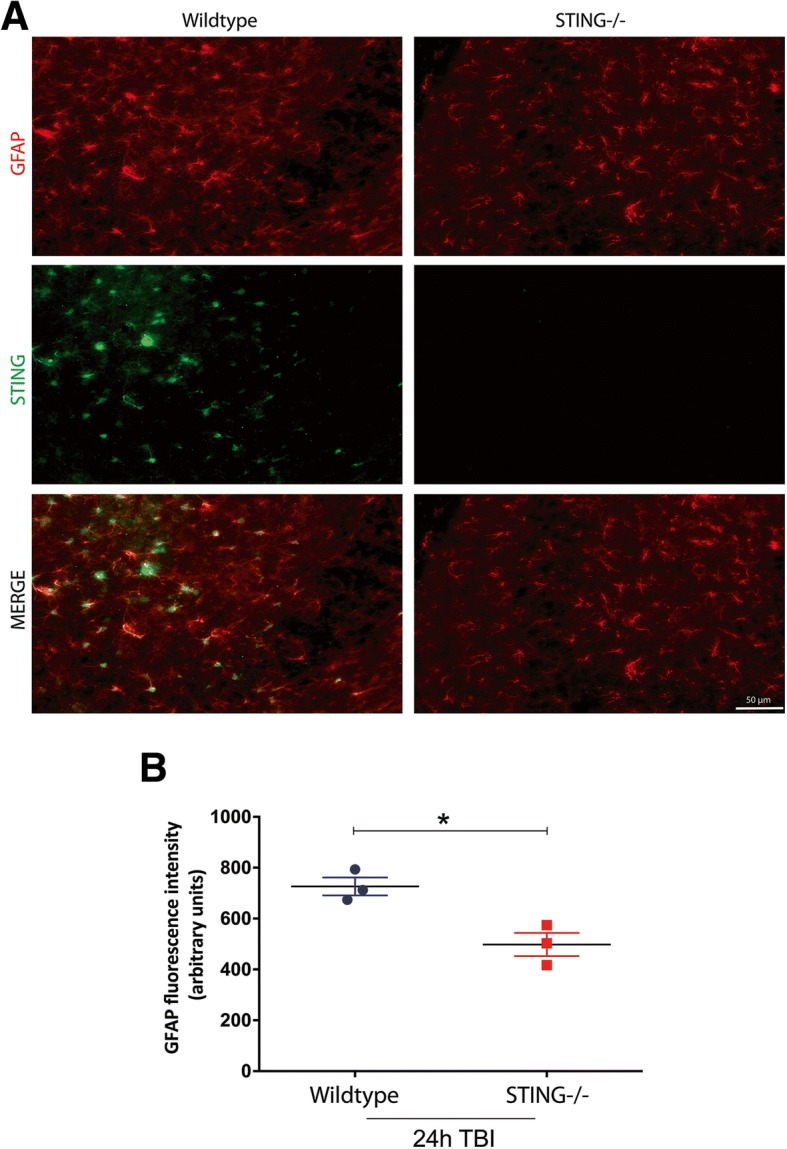
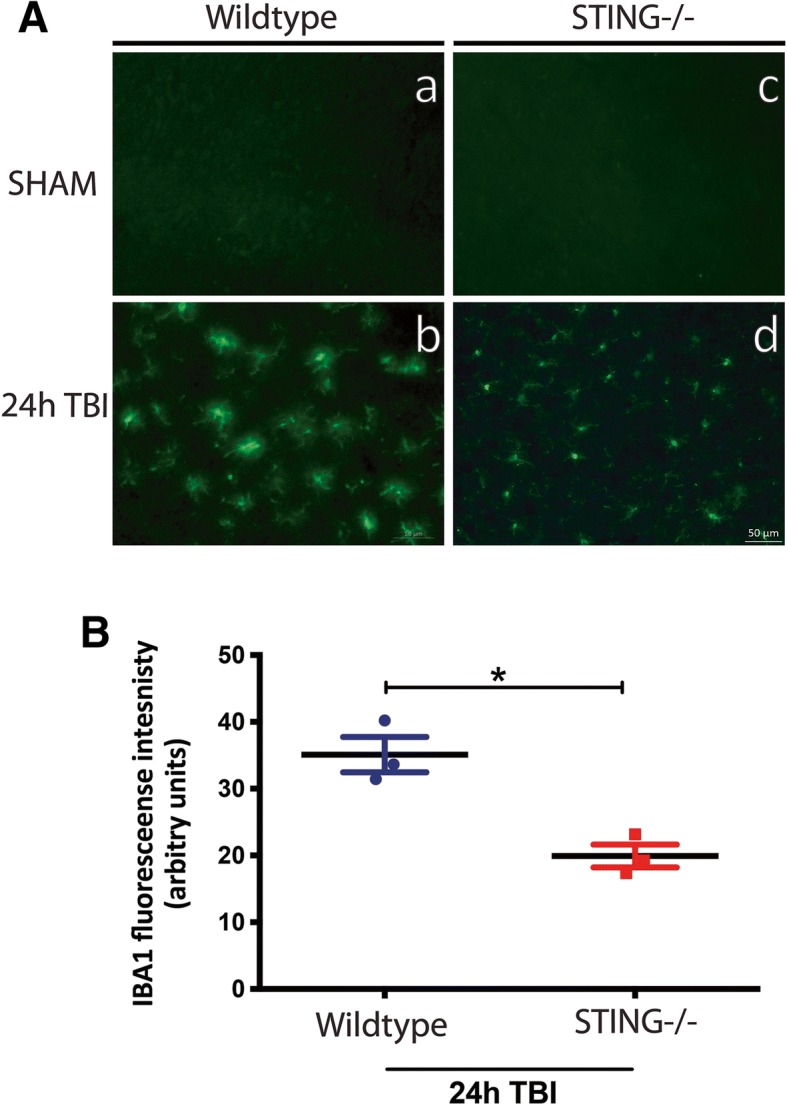
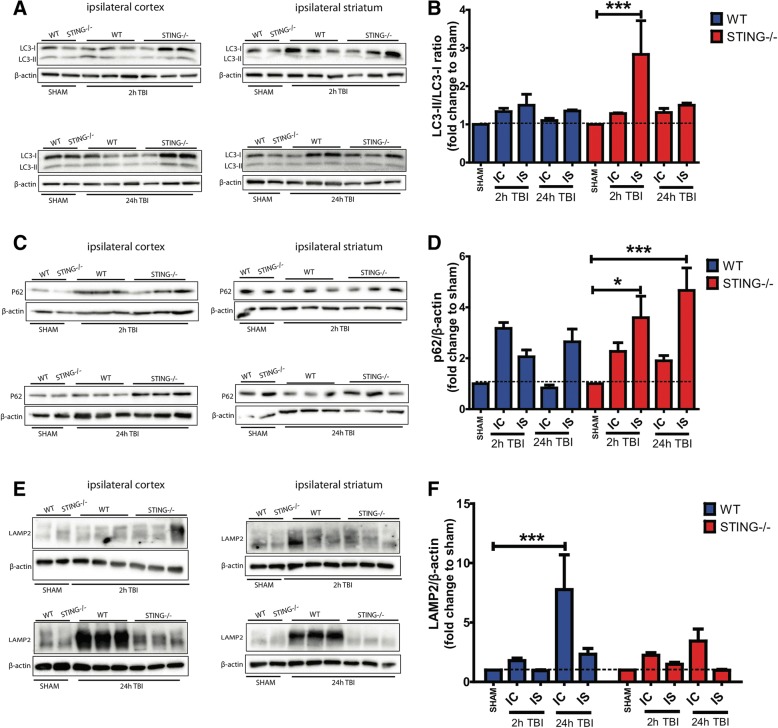
Results: A significant upregulation in STING expression was identified in late trauma human brain samples that was confirmed in wild-type mice at 2 h and 24 h after CCI. This correlated with an elevated pro-inflammatory cytokine profile with increased TNF-α, IL-6, IL-1β and type-I IFN (IFN-α and IFN-β) levels. This expression was suppressed in the STING-/- mice with a smaller lesion volume in the knockout animals at 24 h post CCI. Wild-type mice also displayed increased levels of autophagy markers, LC3-II, p62 and LAMP2 after TBI; however, STING-/- mice showed reduced LAMP2 expression suggesting a role for STING in driving dysfunctional autophagy after TBI.
Conclusion: Our data implicates a detrimental role for STING in mediating the TBI-induced neuroinflammatory response and autophagy dysfunction, potentially identifying a new therapeutic target for reducing cellular damage in TBI.
Keywords: Autophagy; Neuroinflammation; STING; Traumatic brain injury; Type-I interferon.
Conflict of interest statement
Ethics approval and consent to participate
All experiments involving animal surgery were conducted in accordance with University of Melbourne Animal Ethics Committee (#1212477.1). All procedures with human tissues were conducted in accordance with the Australian National Health & Medical Research Council’s National Statement on Ethical Conduct in Human Research (2007), the Victorian Human Tissue Act 1982, the National Code of Ethical Autopsy Practice and the Victorian Government Policies and Practices in Relation to Postmortem.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
