

Adaptation to DNA damage checkpoint in senescent telomerase-negative cells promotes genome instability
- PMID: 30463903
- PMCID: PMC6295172
- DOI: 10.1101/gad.318485.118
Adaptation to DNA damage checkpoint in senescent telomerase-negative cells promotes genome instability
Erratum in
-
Corrigendum: Adaptation to DNA damage checkpoint in senescent telomerase-negative cells promotes genome instability.Genes Dev. 2019 Mar 1;33(5-6):378. doi: 10.1101/gad.324459.119. Genes Dev. 2019. PMID: 30824534 Free PMC article. No abstract available.
Abstract
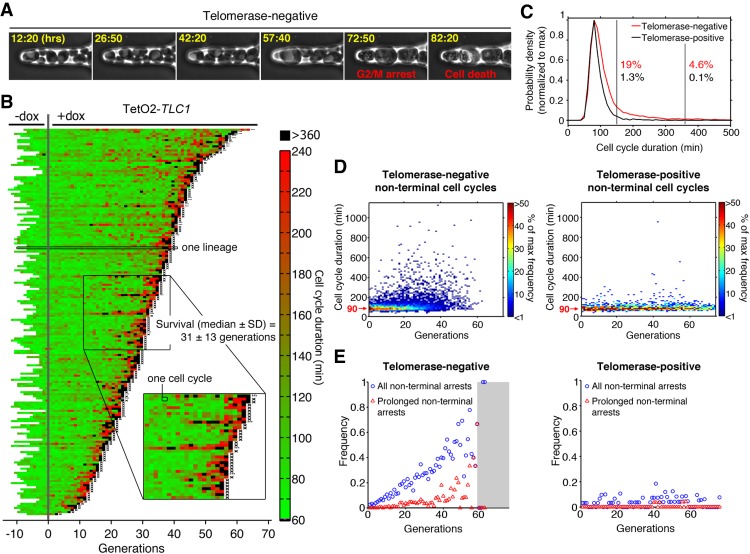
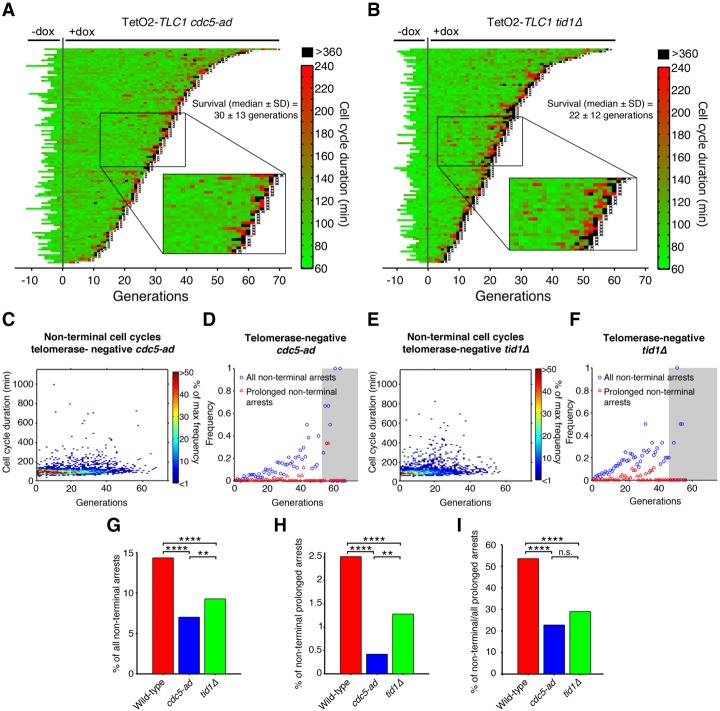
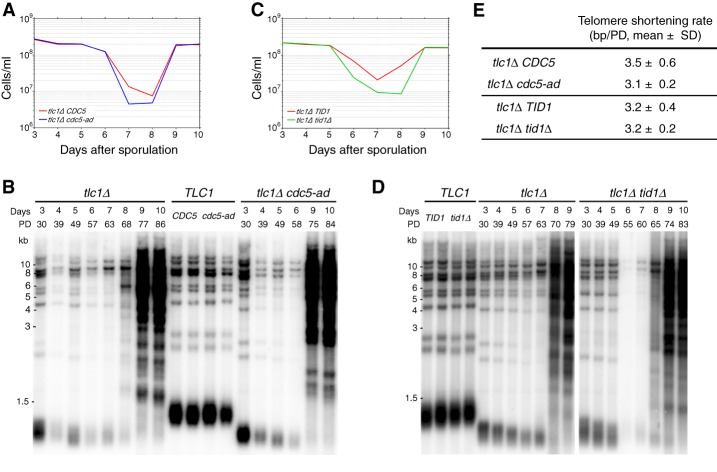
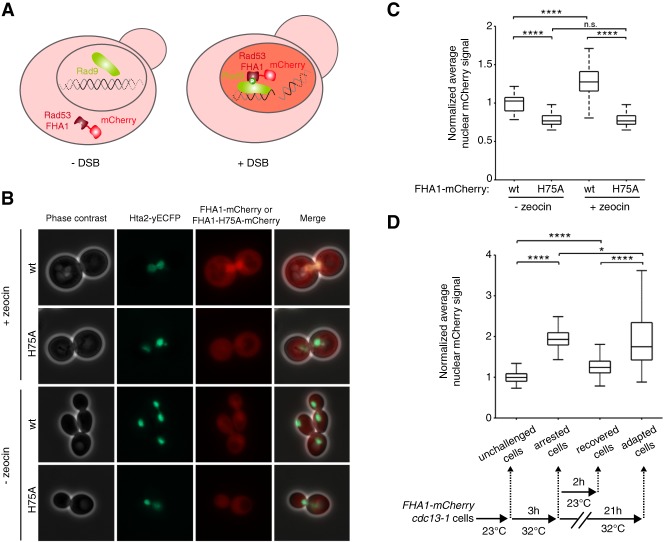
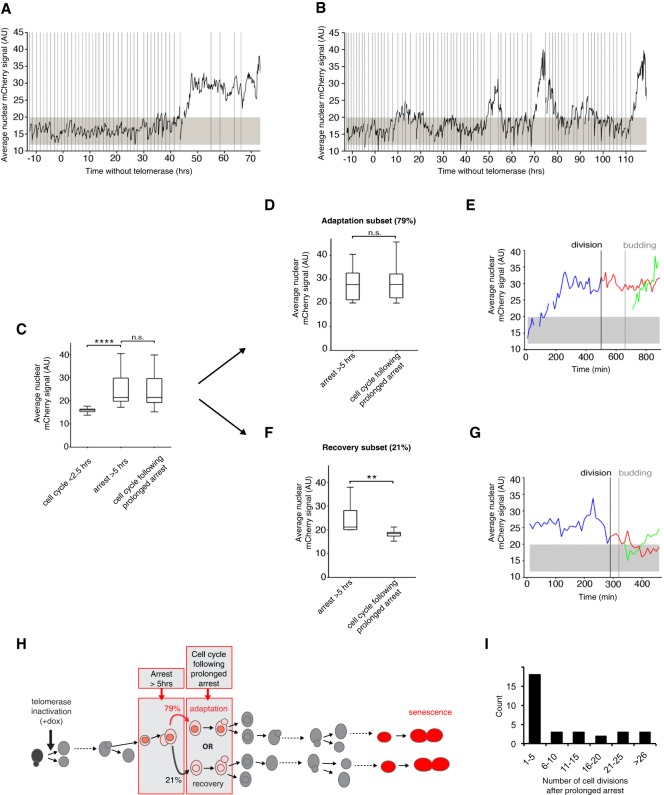
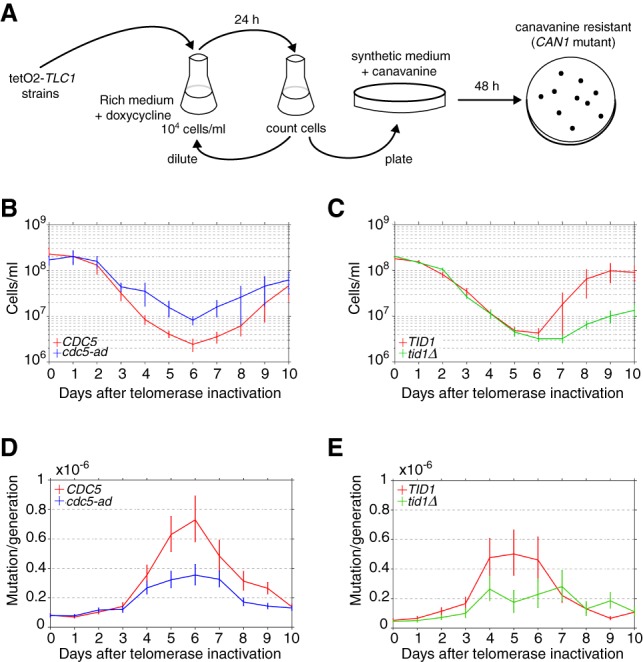
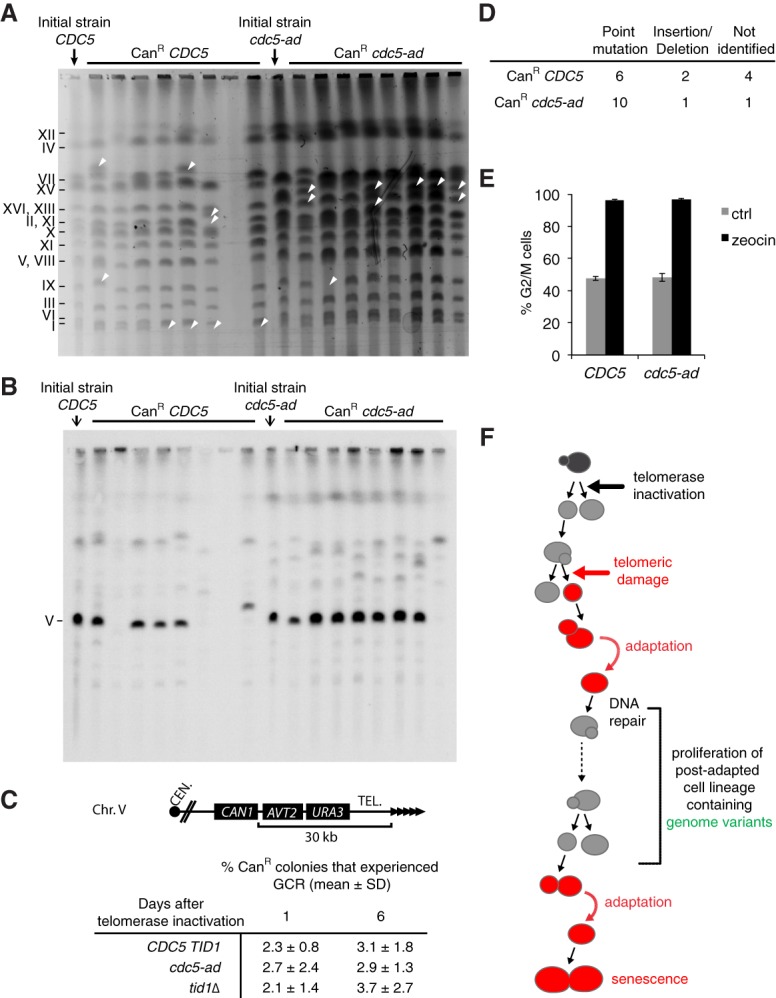
In cells lacking telomerase, telomeres gradually shorten during each cell division to reach a critically short length, permanently activate the DNA damage checkpoint, and trigger replicative senescence. The increase in genome instability that occurs as a consequence may contribute to the early steps of tumorigenesis. However, because of the low frequency of mutations and the heterogeneity of telomere-induced senescence, the timing and mechanisms of genome instability increase remain elusive. Here, to capture early mutation events during replicative senescence, we used a combined microfluidic-based approach and live-cell imaging in yeast. We analyzed DNA damage checkpoint activation in consecutive cell divisions of individual cell lineages in telomerase-negative yeast cells and observed that prolonged checkpoint arrests occurred frequently in telomerase-negative lineages. Cells relied on the adaptation to the DNA damage pathway to bypass the prolonged checkpoint arrests, allowing further cell divisions despite the presence of unrepaired DNA damage. We demonstrate that the adaptation pathway is a major contributor to the genome instability induced during replicative senescence. Therefore, adaptation plays a critical role in shaping the dynamics of genome instability during replicative senescence.
Keywords: Cdc5; DNA damage checkpoint; adaptation; genomic instability; senescence; telomere.
© 2018 Coutelier et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Adaptation in replicative senescence: a risky business.Curr Genet. 2019 Jun;65(3):711-716. doi: 10.1007/s00294-019-00933-7. Epub 2019 Jan 12. Curr Genet. 2019. PMID: 30637477 Review.
-
Tel1/ATM Signaling to the Checkpoint Contributes to Replicative Senescence in the Absence of Telomerase.Genetics. 2019 Oct;213(2):411-429. doi: 10.1534/genetics.119.302391. Epub 2019 Aug 7. Genetics. 2019. PMID: 31391264 Free PMC article.
-
Telomere shortening causes distinct cell division regimes during replicative senescence in Saccharomyces cerevisiae.Cell Biosci. 2021 Oct 9;11(1):180. doi: 10.1186/s13578-021-00693-3. Cell Biosci. 2021. PMID: 34627377 Free PMC article.
-
Two routes to senescence revealed by real-time analysis of telomerase-negative single lineages.Nat Commun. 2015 Jul 9;6:7680. doi: 10.1038/ncomms8680. Nat Commun. 2015. PMID: 26158780 Free PMC article.
-
The many types of heterogeneity in replicative senescence.Yeast. 2019 Nov;36(11):637-648. doi: 10.1002/yea.3433. Epub 2019 Aug 6. Yeast. 2019. PMID: 31306505 Free PMC article. Review.
Cited by
-
Regulation of Mitotic Exit by Cell Cycle Checkpoints: Lessons From Saccharomyces cerevisiae.Genes (Basel). 2020 Feb 12;11(2):195. doi: 10.3390/genes11020195. Genes (Basel). 2020. PMID: 32059558 Free PMC article. Review.
-
Plasma membrane damage limits replicative lifespan in yeast and induces premature senescence in human fibroblasts.Nat Aging. 2024 Mar;4(3):319-335. doi: 10.1038/s43587-024-00575-6. Epub 2024 Feb 22. Nat Aging. 2024. PMID: 38388781 Free PMC article.
-
The Polo kinase Cdc5 is regulated at multiple levels in the adaptation response to telomere dysfunction.Genetics. 2023 Jan 12;223(1):iyac171. doi: 10.1093/genetics/iyac171. Genetics. 2023. PMID: 36342193 Free PMC article.
-
Architecture and evolution of subtelomeres in the unicellular green alga Chlamydomonas reinhardtii.Nucleic Acids Res. 2021 Jul 21;49(13):7571-7587. doi: 10.1093/nar/gkab534. Nucleic Acids Res. 2021. PMID: 34165564 Free PMC article.
-
Comparative analysis of genome-wide transcriptional responses to continuous heat stress in Pleurotus tuoliensis.AMB Express. 2023 Nov 2;13(1):121. doi: 10.1186/s13568-023-01630-y. AMB Express. 2023. PMID: 37917251 Free PMC article.
References
-
- Artandi SE, DePinho RA. 2000. A critical role for telomeres in suppressing and facilitating carcinogenesis. Curr Opin Genet Dev 10: 39–46. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases