

Integrative epigenetic taxonomy of primary prostate cancer
- PMID: 30464211
- PMCID: PMC6249266
- DOI: 10.1038/s41467-018-07270-2
Integrative epigenetic taxonomy of primary prostate cancer
Abstract
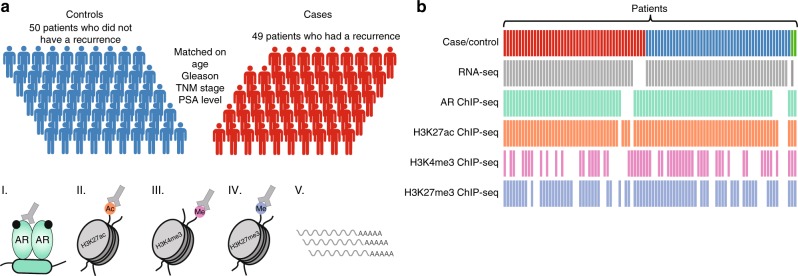
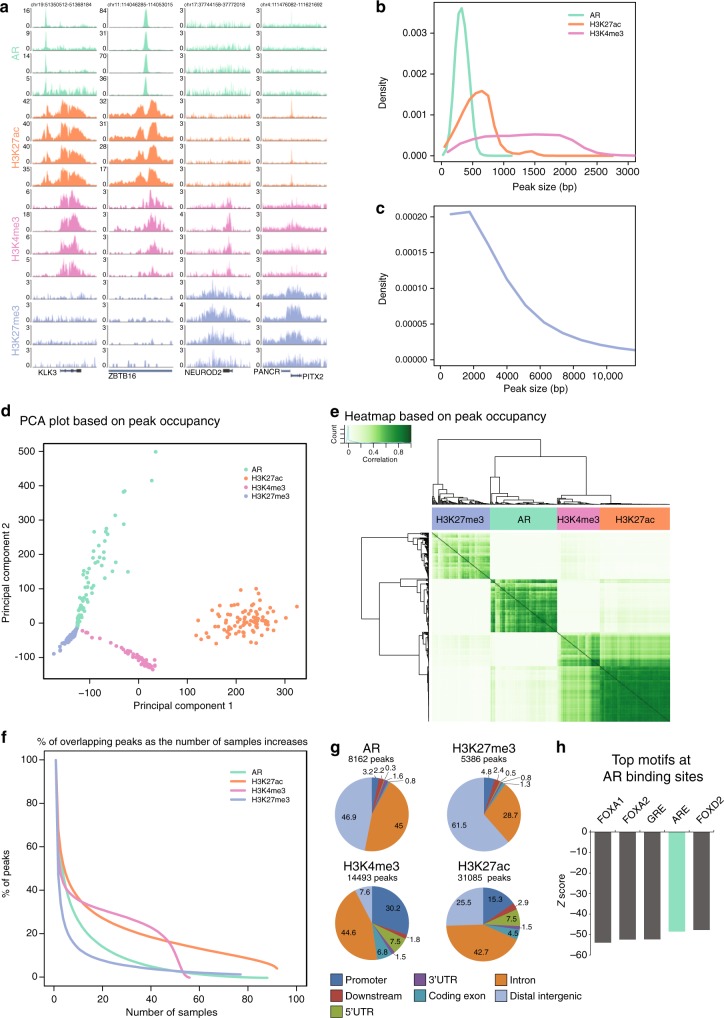
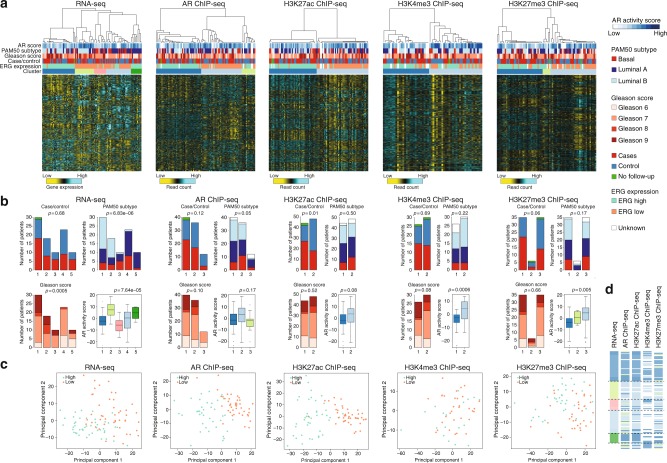
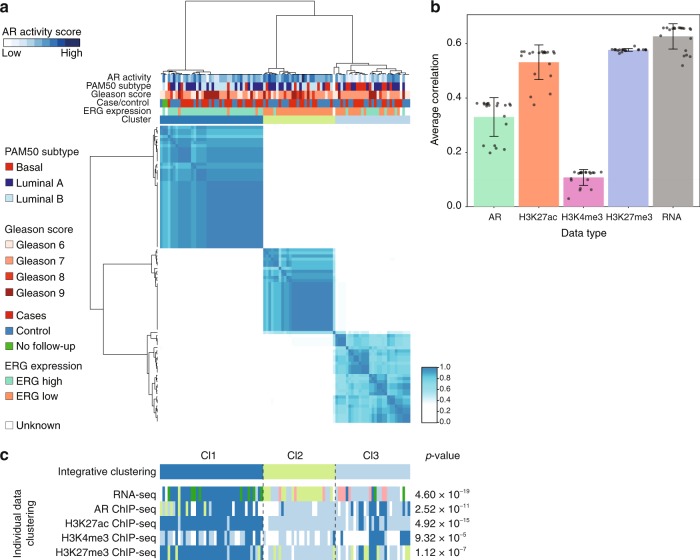
The Androgen Receptor (AR) is the key-driving transcription factor in prostate cancer, tightly controlled by epigenetic regulation. To date, most epigenetic profiling has been performed in cell lines or limited tissue samples. Here, to comprehensively study the epigenetic landscape, we perform RNA-seq with ChIP-seq for AR and histone modification marks (H3K27ac, H3K4me3, H3K27me3) in 100 primary prostate carcinomas. Integrative molecular subtyping of the five data streams revealed three major subtypes of which two were clearly TMPRSS2-ERG dictated. Importantly, we identify a third subtype with low chromatin binding and activity of AR, but with high activity of FGF and WNT signaling. While positive for neuroendocrine-hallmark genes, these tumors were copy number-neutral with low mutational burden, significantly depleted for genes characteristic of poor-outcome associated luminal B-subtype. We present a unique resource on transcriptional and epigenetic control in prostate cancer, revealing tight control of gene regulation differentially dictated by AR over three subtypes.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Roehl KA, Han M, Ramos CG, Antenor JA, Catalona WJ. Cancer progression and survival rates following anatomical radical retropubic prostatectomy in 3,478 consecutive patients: long-term results. J. Urol. 2004;172:910–914. doi: 10.1097/01.ju.0000134888.22332.bb. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
