

Amphioxus functional genomics and the origins of vertebrate gene regulation
- PMID: 30464347
- PMCID: PMC6292497
- DOI: 10.1038/s41586-018-0734-6
Amphioxus functional genomics and the origins of vertebrate gene regulation
Abstract
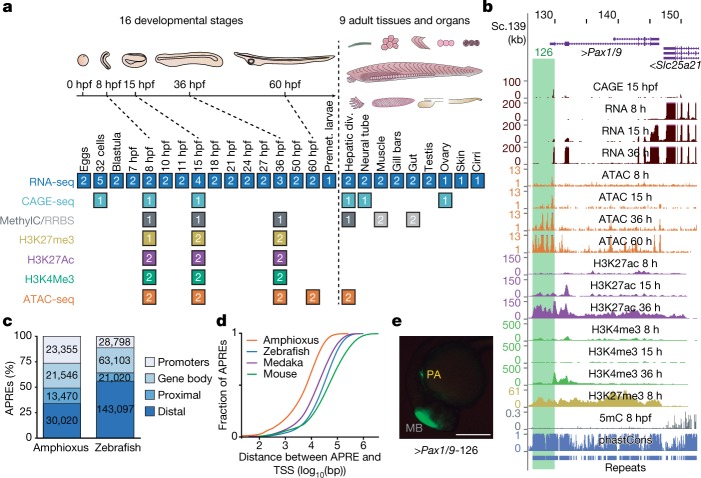
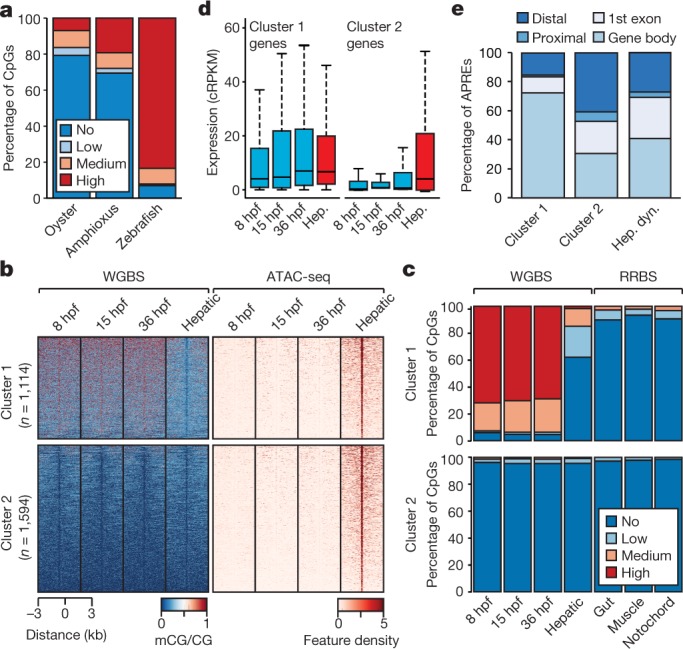
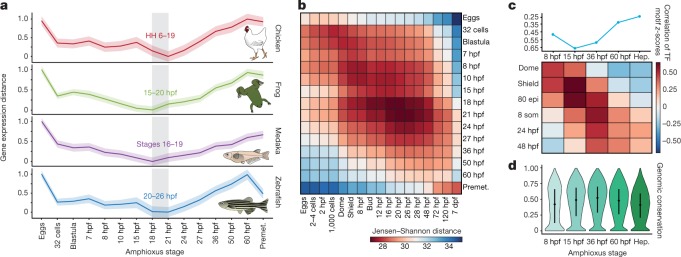
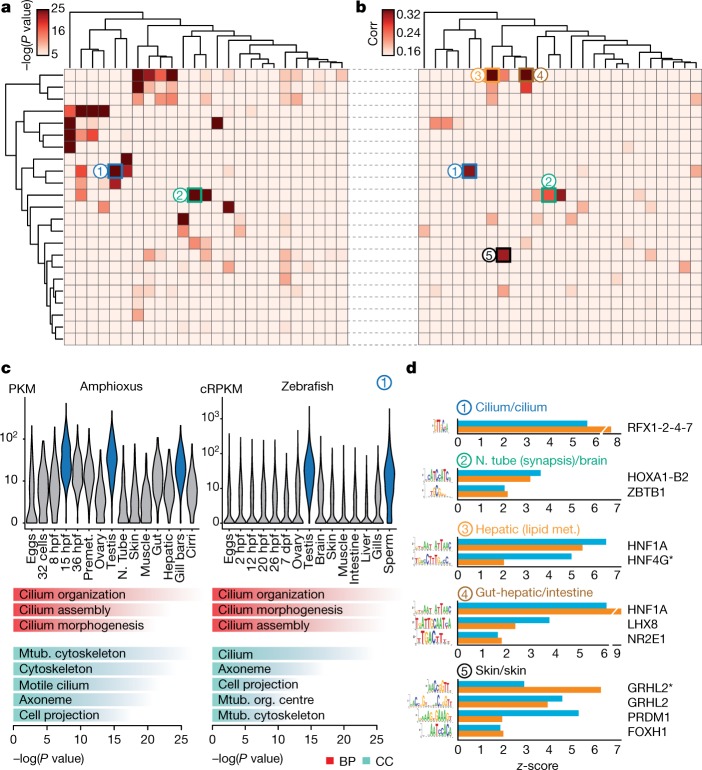
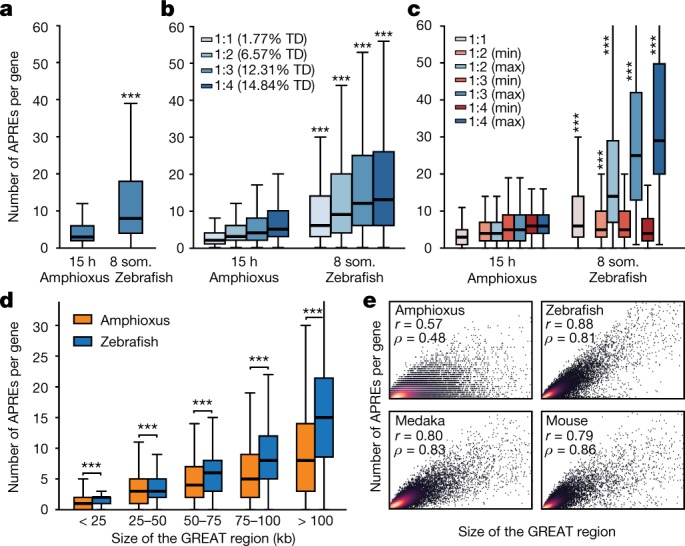
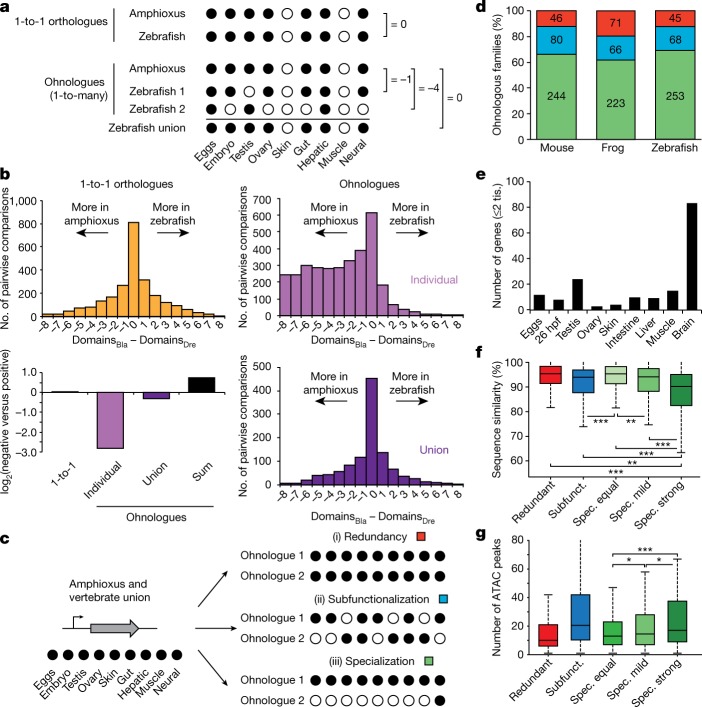
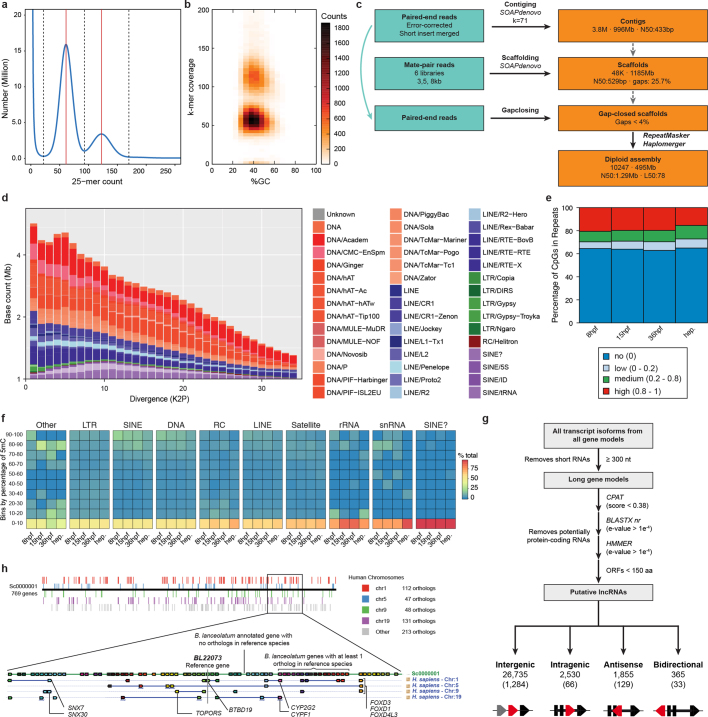
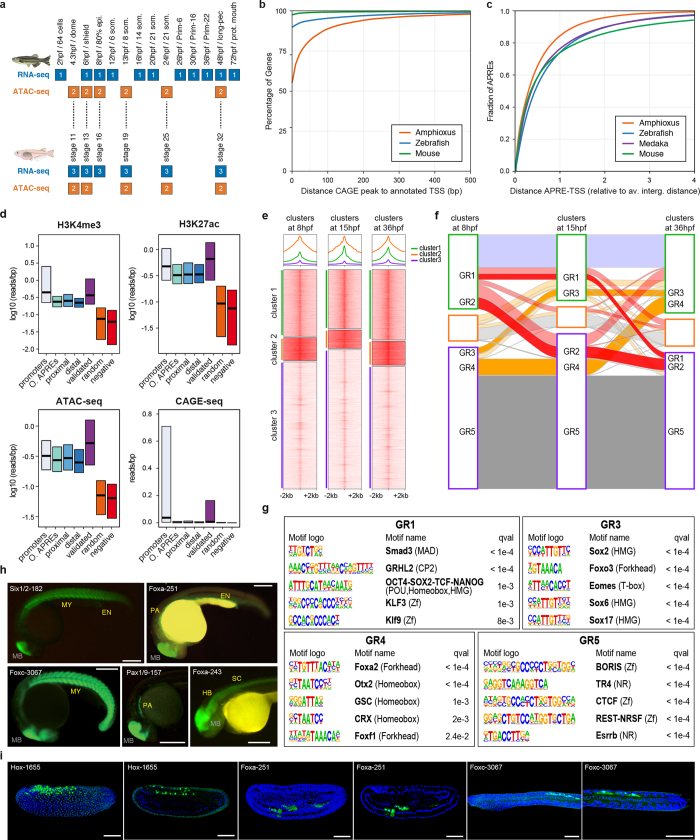
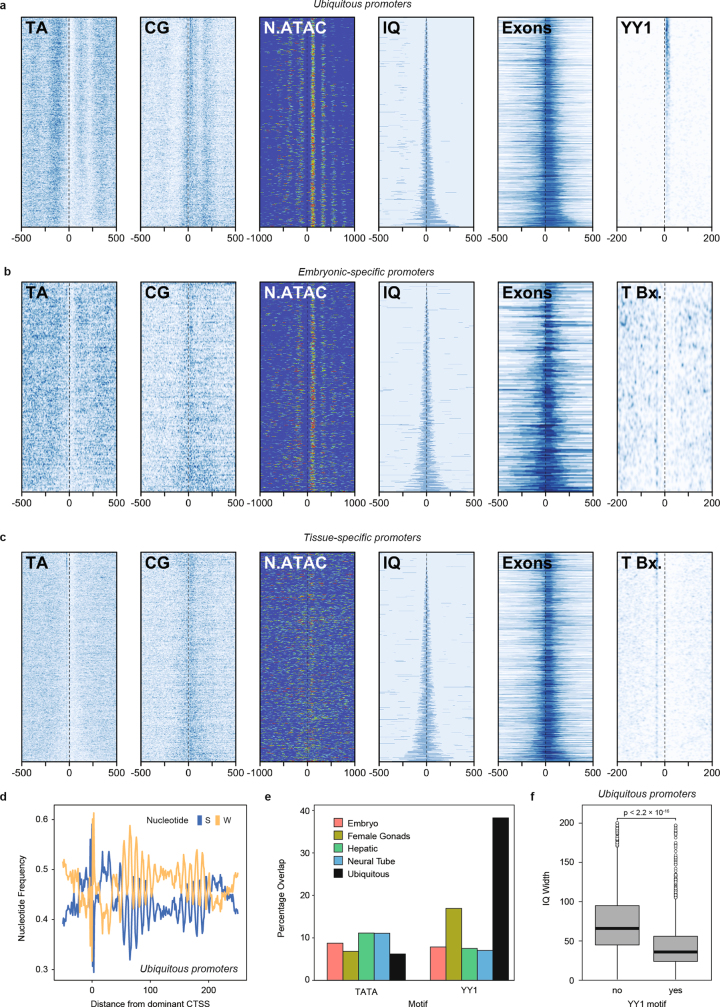
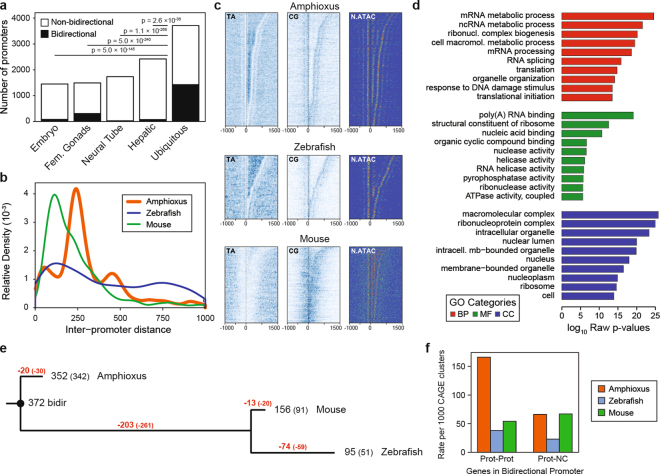
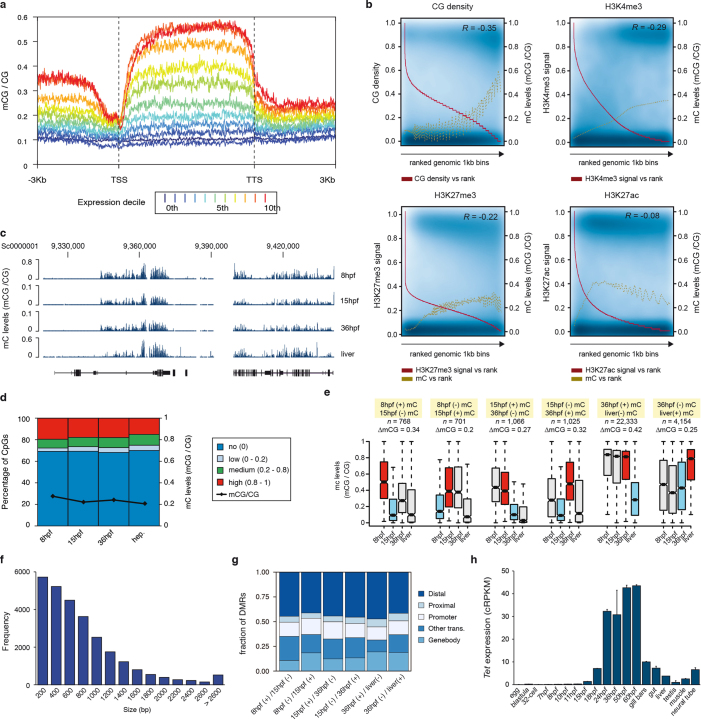
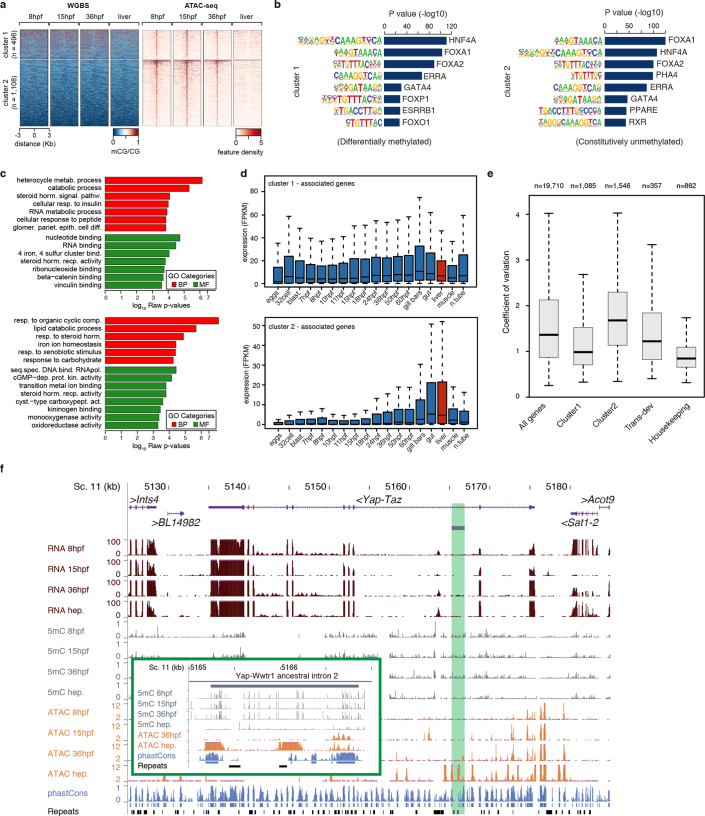
Vertebrates have greatly elaborated the basic chordate body plan and evolved highly distinctive genomes that have been sculpted by two whole-genome duplications. Here we sequence the genome of the Mediterranean amphioxus (Branchiostoma lanceolatum) and characterize DNA methylation, chromatin accessibility, histone modifications and transcriptomes across multiple developmental stages and adult tissues to investigate the evolution of the regulation of the chordate genome. Comparisons with vertebrates identify an intermediate stage in the evolution of differentially methylated enhancers, and a high conservation of gene expression and its cis-regulatory logic between amphioxus and vertebrates that occurs maximally at an earlier mid-embryonic phylotypic period. We analyse regulatory evolution after whole-genome duplications, and find that-in vertebrates-over 80% of broadly expressed gene families with multiple paralogues derived from whole-genome duplications have members that restricted their ancestral expression, and underwent specialization rather than subfunctionalization. Counter-intuitively, paralogues that restricted their expression increased the complexity of their regulatory landscapes. These data pave the way for a better understanding of the regulatory principles that underlie key vertebrate innovations.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
