

Targeting intermediary metabolism enhances the efficacy of BH3 mimetic therapy in hematologic malignancies
- PMID: 30467206
- PMCID: PMC6518917
- DOI: 10.3324/haematol.2018.204701
Targeting intermediary metabolism enhances the efficacy of BH3 mimetic therapy in hematologic malignancies
Abstract
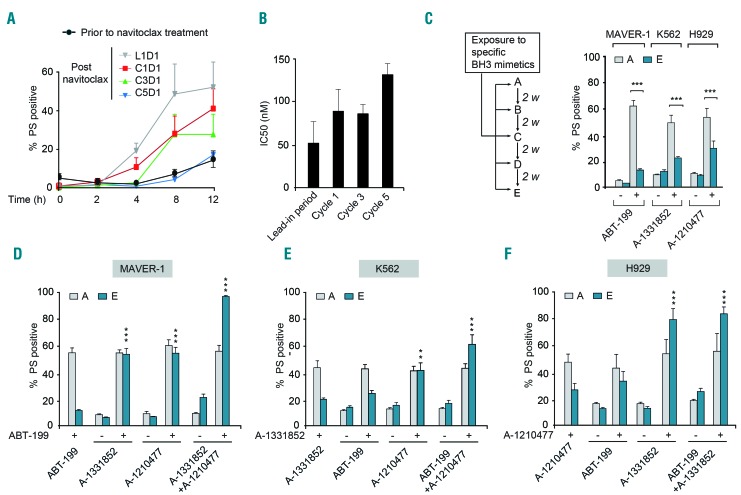
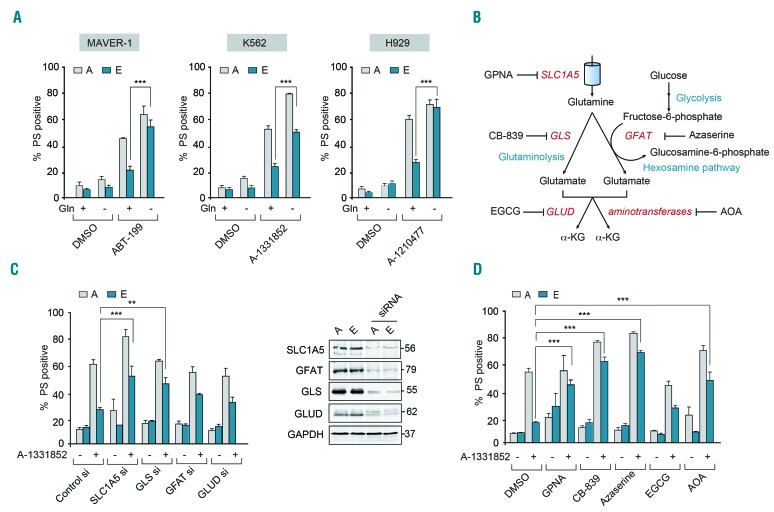
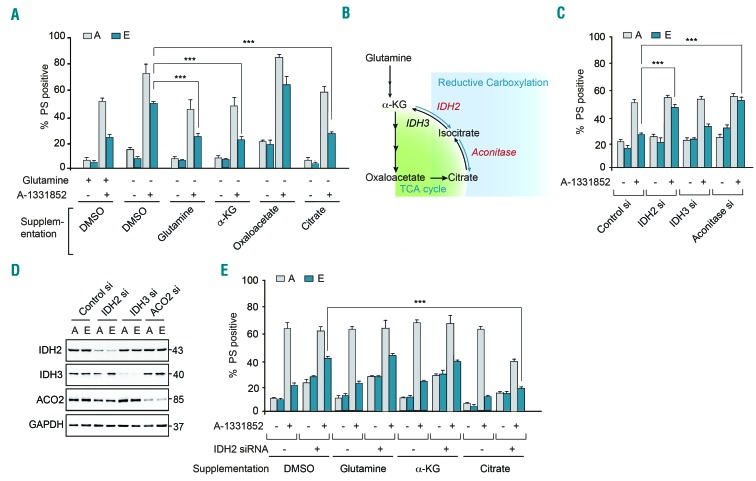
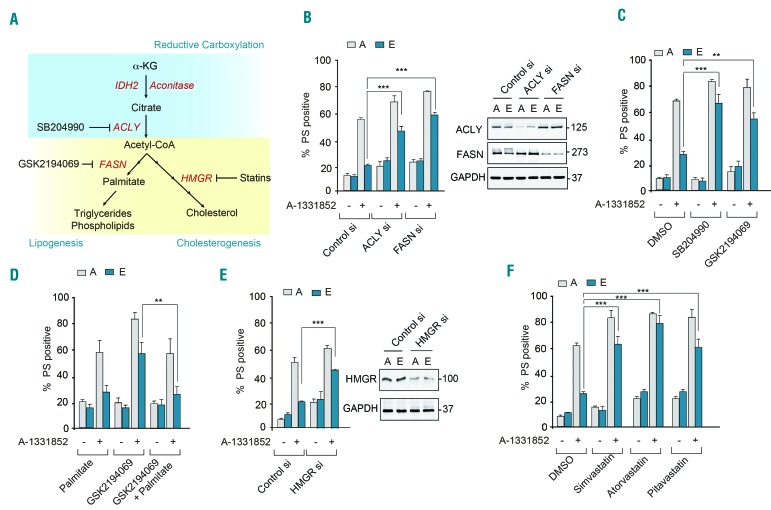
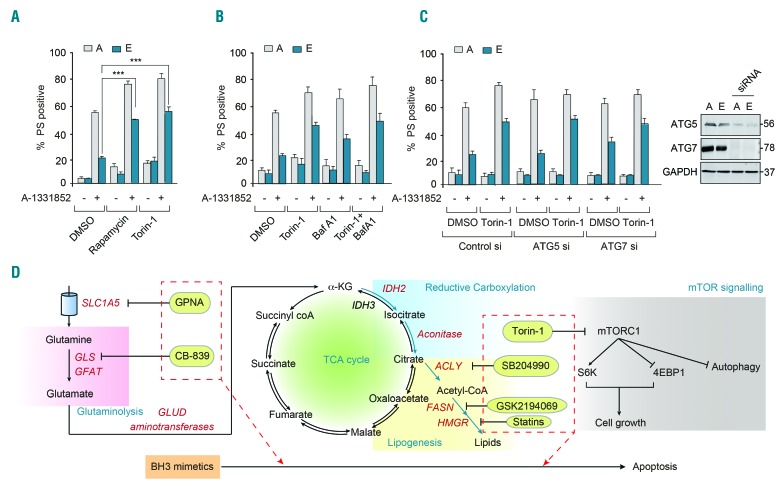
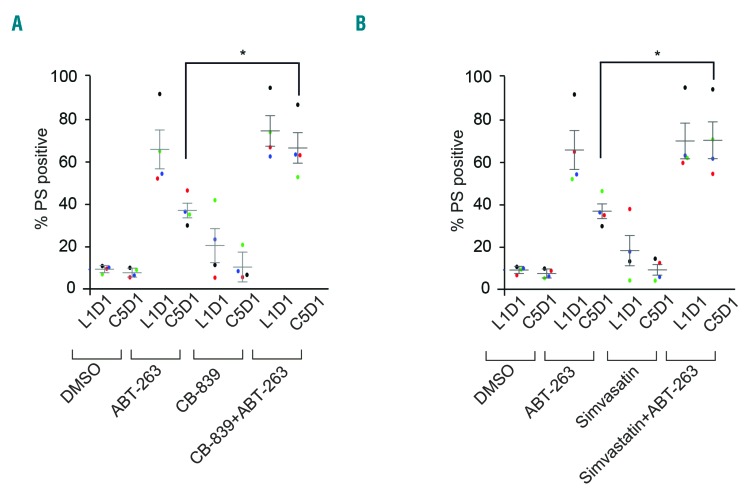
BH3 mimetics are novel targeted drugs with remarkable specificity, potency and enormous potential to improve cancer therapy. However, acquired resistance is an emerging problem. We report the rapid development of resistance in chronic lymphocytic leukemia cells isolated from patients exposed to increasing doses of navitoclax (ABT-263), a BH3 mimetic. To mimic such rapid development of chemoresistance, we developed simple resistance models to three different BH3 mimetics, targeting BCL-2 (ABT-199), BCL-XL (A-1331852) or MCL-1 (A-1210477), in relevant hematologic cancer cell lines. In these models, resistance could not be attributed to either consistent changes in expression levels of the anti-apoptotic proteins or interactions among different pro- and anti-apoptotic BCL-2 family members. Using genetic silencing, pharmacological inhibition and metabolic supplementation, we found that targeting glutamine uptake and its downstream signaling pathways, namely glutaminolysis, reductive carboxylation, lipogenesis, cholesterogenesis and mammalian target of rapamycin signaling resulted in marked sensitization of the chemoresistant cells to BH3 mimetic-mediated apoptosis. Furthermore, our findings highlight the possibility of repurposing widely used drugs, such as statins, to target intermediary metabolism and improve the efficacy of BH3 mimetic therapy.
Copyright© 2019 Ferrata Storti Foundation.
Figures
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. - PubMed
-
- Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9): 3421–3428. - PubMed
-
- Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. - PubMed
-
- Leverson JD, Phillips DC, Mitten MJ, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7(279):279ra40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
