

Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases
- PMID: 30467385
- PMCID: PMC6342007
- DOI: 10.1038/s41583-018-0093-1
Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases
Abstract
Apoptosis is crucial for the normal development of the nervous system, whereas neurons in the adult CNS are relatively resistant to this form of cell death. However, under pathological conditions, upregulation of death receptor family ligands, such as tumour necrosis factor (TNF), can sensitize cells in the CNS to apoptosis and a form of regulated necrotic cell death known as necroptosis that is mediated by receptor-interacting protein kinase 1 (RIPK1), RIPK3 and mixed lineage kinase domain-like protein (MLKL). Necroptosis promotes further cell death and neuroinflammation in the pathogenesis of several neurodegenerative diseases, including multiple sclerosis, amyotrophic lateral sclerosis, Parkinson disease and Alzheimer disease. In this Review, we outline the evidence implicating necroptosis in these neurological diseases and suggest that targeting RIPK1 might help to inhibit multiple cell death pathways and ameliorate neuroinflammation.
Conflict of interest statement
Competing interests
J.Y. is a consultant of Denali Therapeutics. P.A. declares no competing interests. D.O. is an employee of Sanofi.
Figures
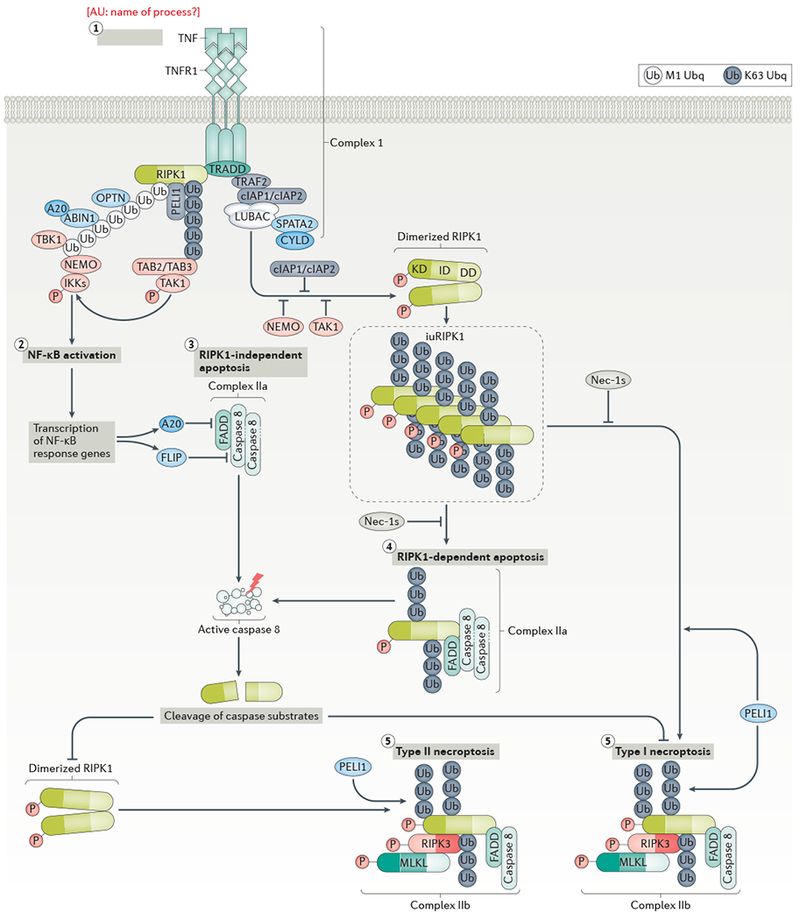
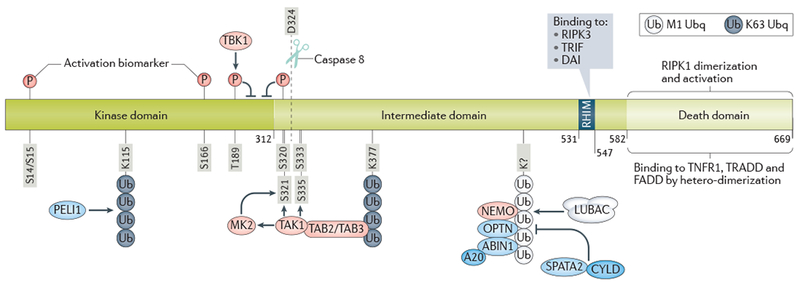
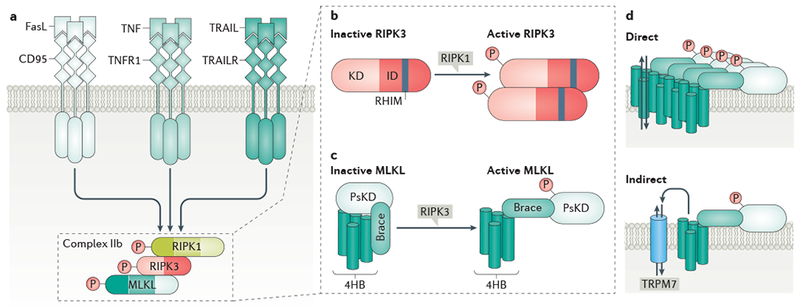
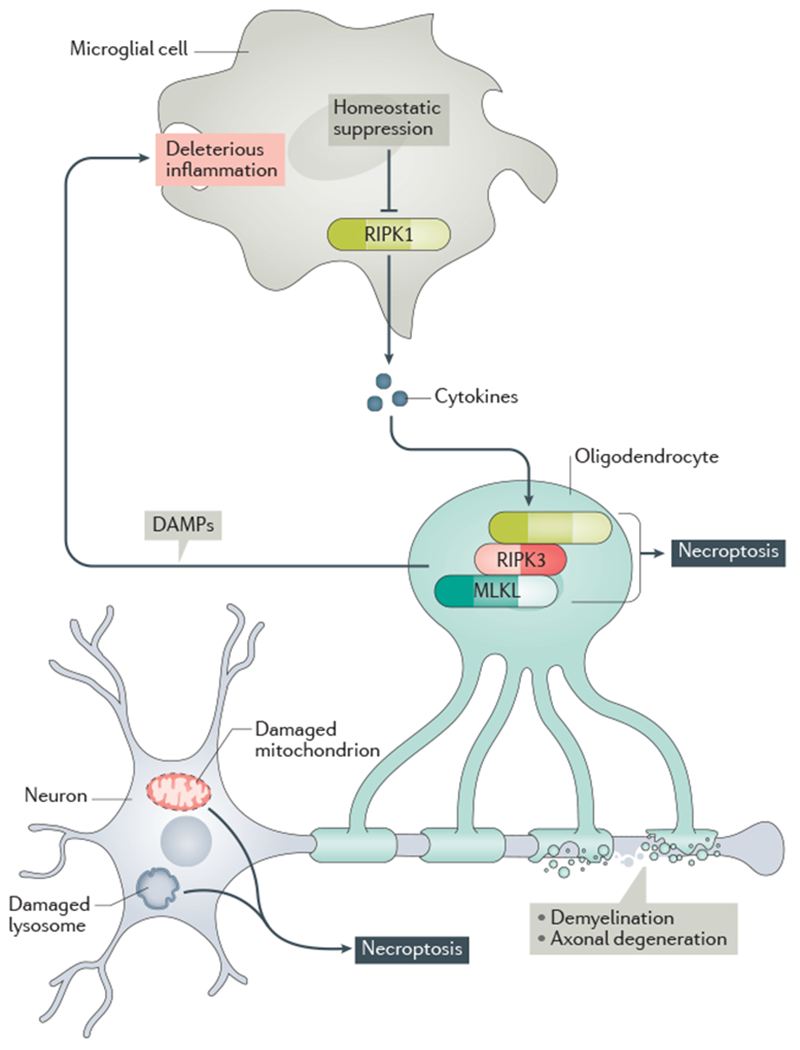
References
-
- Degterev A et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1, 112–119 (2005). - PubMed
-
This paper provided the first definition of necroptosis and insights into the functional role of necroptosis in acute neurological injuries.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
