

Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy
- PMID: 30467425
- PMCID: PMC6250721
- DOI: 10.1038/s41467-018-07261-3
Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy
Abstract
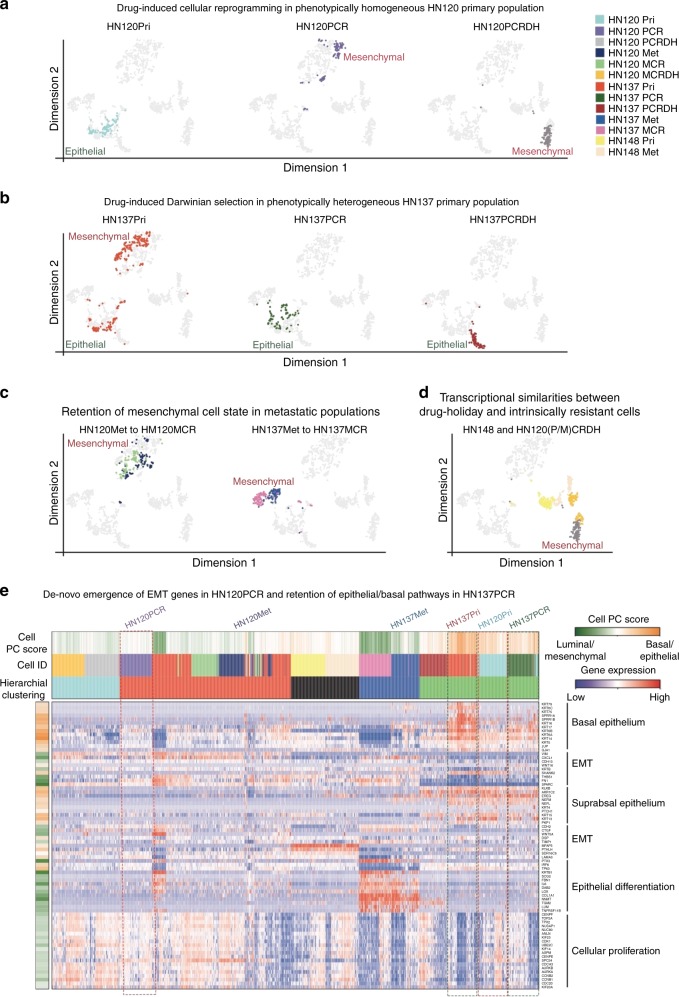
Chemo-resistance is one of the major causes of cancer-related deaths. Here we used single-cell transcriptomics to investigate divergent modes of chemo-resistance in tumor cells. We observed that higher degree of phenotypic intra-tumor heterogeneity (ITH) favors selection of pre-existing drug-resistant cells, whereas phenotypically homogeneous cells engage covert epigenetic mechanisms to trans-differentiate under drug-selection. This adaptation was driven by selection-induced gain of H3K27ac marks on bivalently poised resistance-associated chromatin, and therefore not expressed in the treatment-naïve setting. Mechanistic interrogation of this phenomenon revealed that drug-induced adaptation was acquired upon the loss of stem factor SOX2, and a concomitant gain of SOX9. Strikingly we observed an enrichment of SOX9 at drug-induced H3K27ac sites, suggesting that tumor evolution could be driven by stem cell-switch-mediated epigenetic plasticity. Importantly, JQ1 mediated inhibition of BRD4 could reverse drug-induced adaptation. These results provide mechanistic insights into the modes of therapy-induced cellular plasticity and underscore the use of epigenetic inhibitors in targeting tumor evolution.
Conflict of interest statement
The authors declare no competing interests.
Figures






Comment in
-
Hiding in Plain Sight: Epigenetic Plasticity in Drug-Induced Tumor Evolution.Epigenet Insights. 2019 Aug 14;12:2516865719870760. doi: 10.1177/2516865719870760. eCollection 2019. Epigenet Insights. 2019. PMID: 31453434 Free PMC article.
-
Variant calling from scRNA-seq data allows the assessment of cellular identity in patient-derived cell lines.Nat Commun. 2022 May 12;13(1):2718. doi: 10.1038/s41467-022-30230-w. Nat Commun. 2022. PMID: 35551450 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
