

Testin protects against cardiac hypertrophy by targeting a calcineurin-dependent signalling pathway
- PMID: 30467953
- PMCID: PMC6307772
- DOI: 10.1111/jcmm.13934
Testin protects against cardiac hypertrophy by targeting a calcineurin-dependent signalling pathway
Abstract
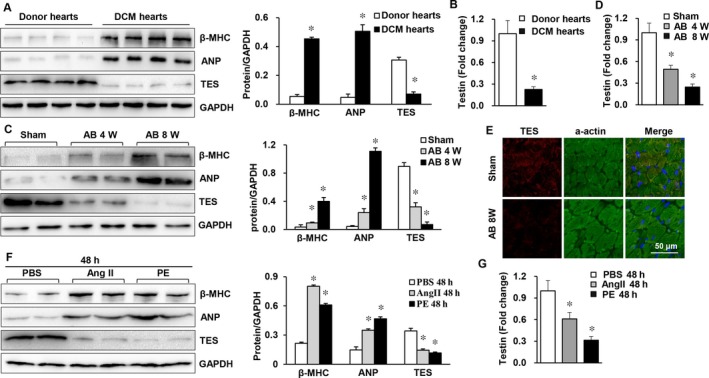
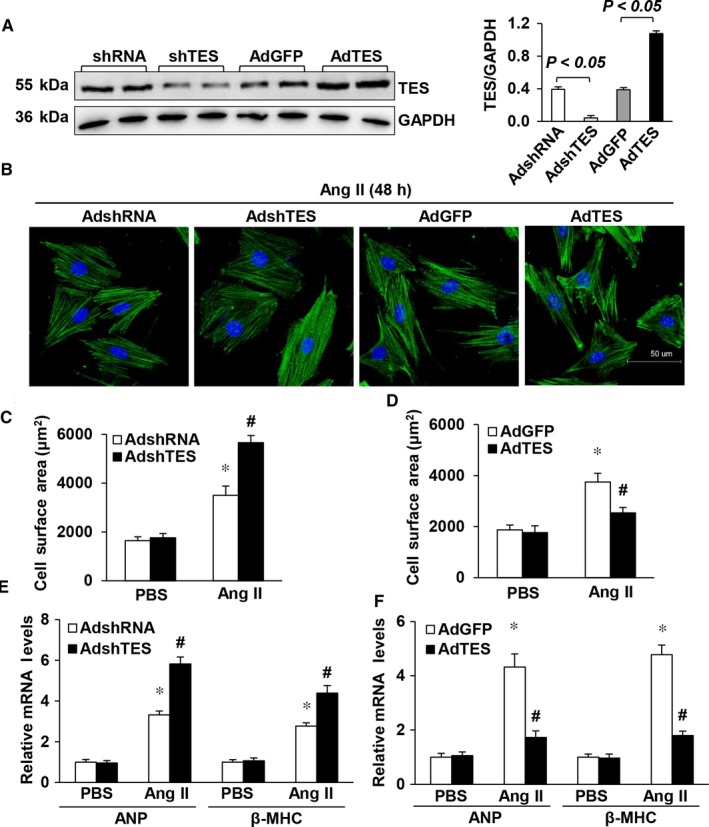
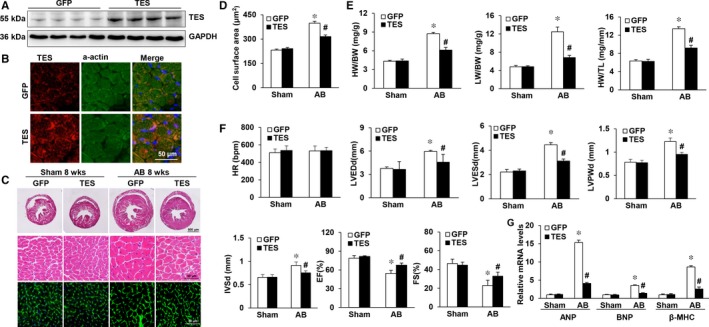
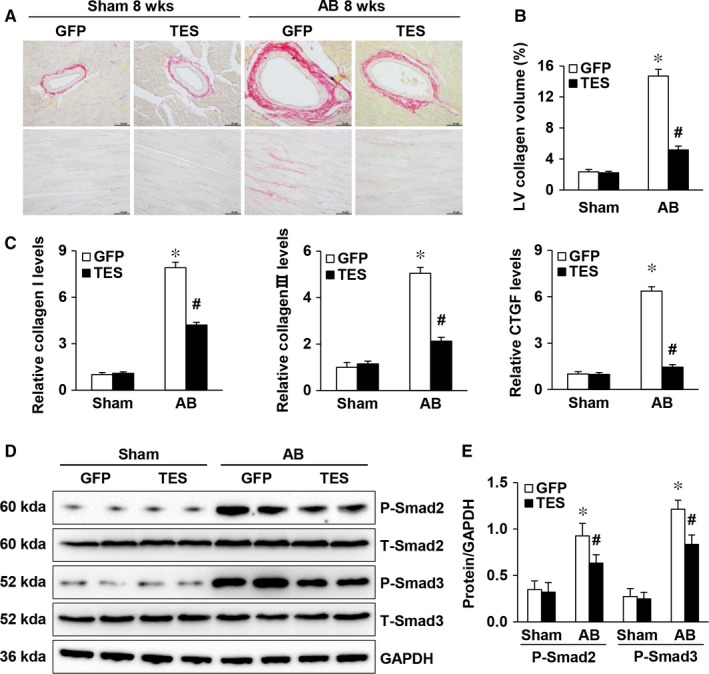
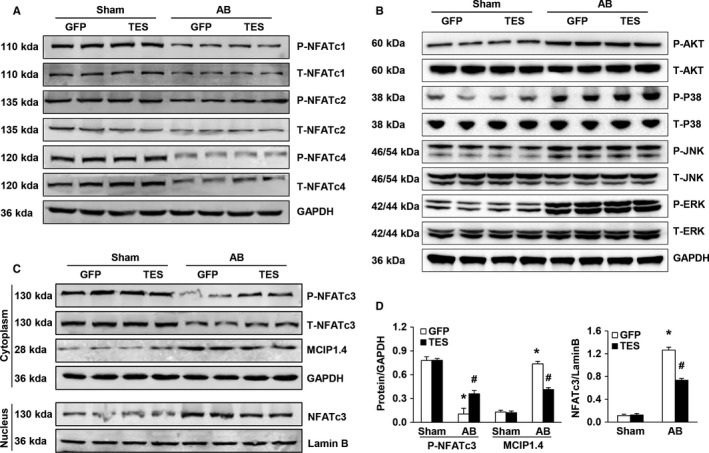
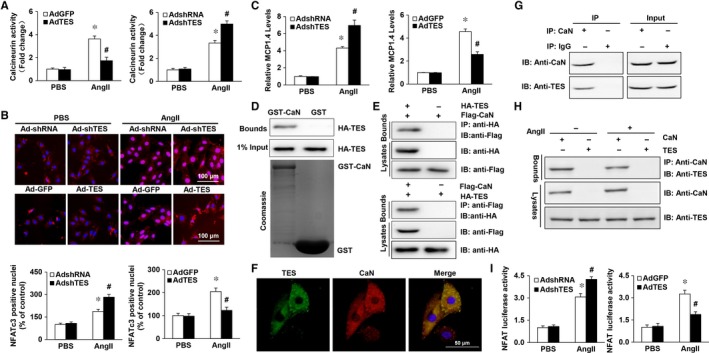
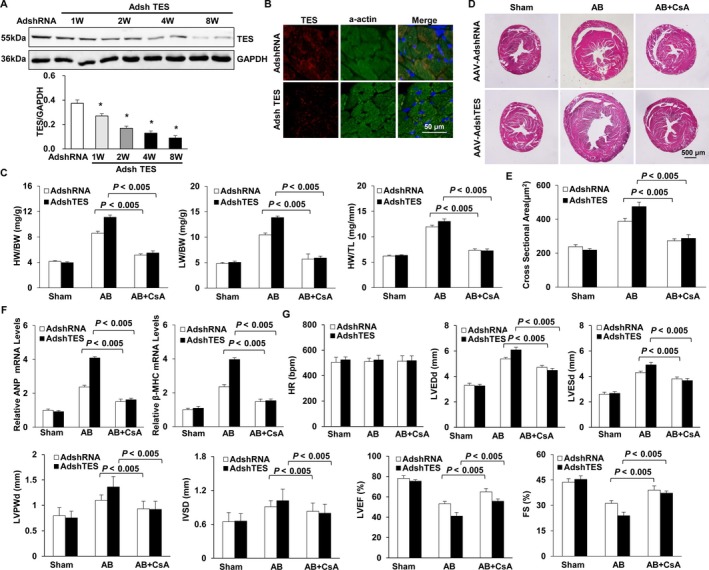
Multiple organs express testin (TES), including the heart. Nevertheless, current understanding of the influence of TES on cardiovascular diseases, especially on cardiac hypertrophy and its etiology, is insufficient. This study investigated the influence of TES on cardiac hypertrophy and its etiology. Murine models with excessive TES expression specific to the heart were constructed with an adeno-associated virus expression system. Cardiac hypertrophy was stimulated through aortic banding (AB). The severity of cardiac hypertrophy was evaluated through molecular, echocardiographic, pathological, and hemodynamic examination. The findings of our study revealed that TES expression was remarkably suppressed not only in failing human hearts but also in mouse hearts with cardiac hypertrophy. It was discovered that excessive TES expression driven by an adeno-associated viral vector noticeably inhibited hypertrophy triggered by angiotensin II (Ang II) in cultivated cardiomyocytes from newborn rats. It was also revealed that TES knockdown via AdshTES caused the reverse phenotype in cardiomyocytes. Furthermore, it was proved that excessive TES expression attenuated the ventricular dilation, cardiac hypertrophy, dysfunction, and fibrosis triggered by AB in mice. It was discovered that TES directly interacted with calcineurin and suppressed its downstream signalling pathway. Moreover, the inactivation of calcineurin with cyclosporin A greatly offset the exacerbated hypertrophic response triggered by AB in TES knockdown mice. Overall, the findings of our study suggest that TES serves as a crucial regulator of the hypertrophic reaction by hindering the calcineurin-dependent pathway in the heart.
Keywords: calcineurin; cardiac hypertrophy; signalling pathway; testin.
© 2018 The Authors. Journal of Cellular and Molecular Medicine published by John Wiley & Sons Ltd and Foundation for Cellular and Molecular Medicine.
Figures
References
-
- Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245‐262. - PubMed
-
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589‐600. - PubMed
-
- Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327‐334. - PubMed
-
- Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242‐255. - PubMed
-
- Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89:1401‐1438. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
