

Transcriptome Deconvolution of Heterogeneous Tumor Samples with Immune Infiltration
- PMID: 30469014
- PMCID: PMC6249353
- DOI: 10.1016/j.isci.2018.10.028
Transcriptome Deconvolution of Heterogeneous Tumor Samples with Immune Infiltration
Abstract
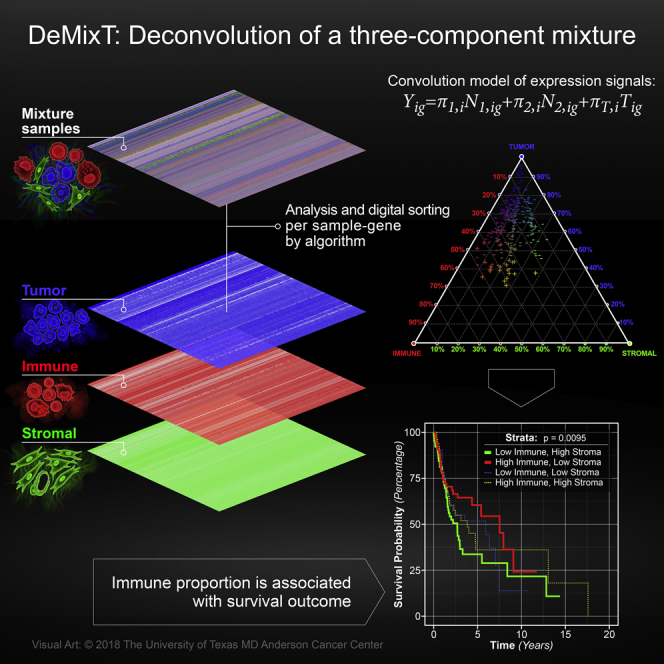
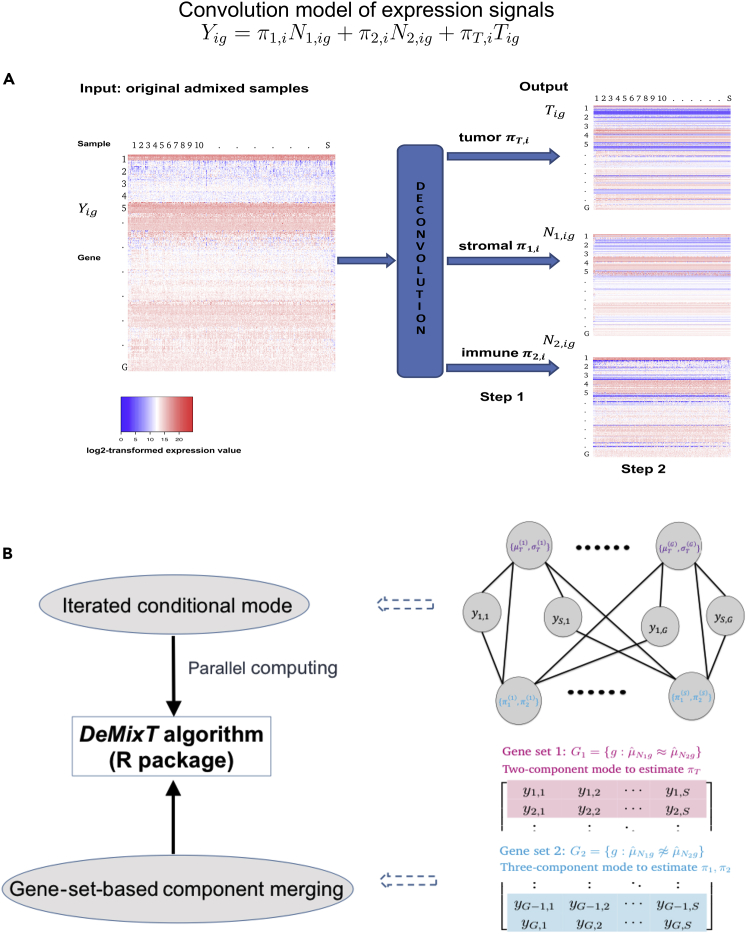
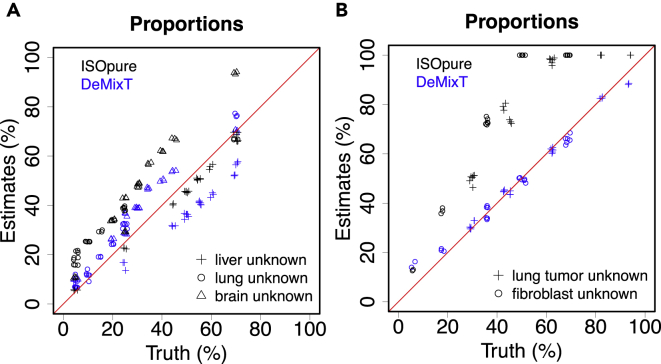
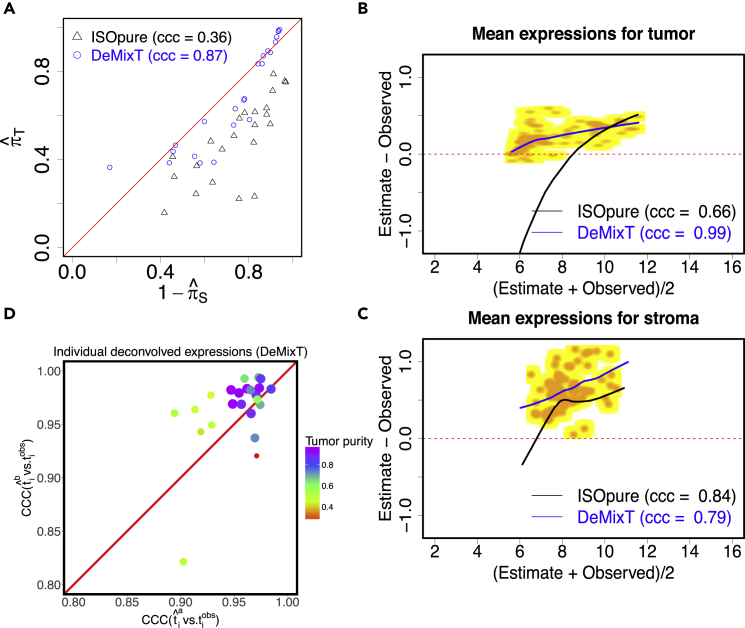
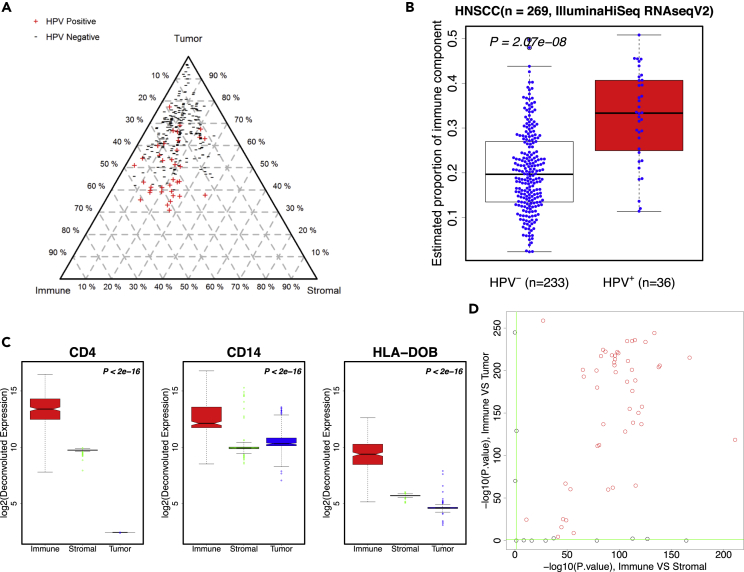
Transcriptome deconvolution in cancer and other heterogeneous tissues remains challenging. Available methods lack the ability to estimate both component-specific proportions and expression profiles for individual samples. We present DeMixT, a new tool to deconvolve high-dimensional data from mixtures of more than two components. DeMixT implements an iterated conditional mode algorithm and a novel gene-set-based component merging approach to improve accuracy. In a series of experimental validation studies and application to TCGA data, DeMixT showed high accuracy. Improved deconvolution is an important step toward linking tumor transcriptomic data with clinical outcomes. An R package, scripts, and data are available: https://github.com/wwylab/DeMixTallmaterials.
Keywords: Cancer; Computational Bioinformatics; Transcriptomics.
Published by Elsevier Inc.
Figures
References
-
- Gong T., Szustakowski J.D. DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics. 2013;29:1083–1085. - PubMed
-
- Besag J. On the statistical analysis of dirty pictures. J. R. Stat. Soc. Series B Stat. Methodol. 1986;48:259–302.
-
- Dave S.S., Wright G., Tan B., Rosenwald A., Gascoyne R.D., Chan W.C., Fisher R.I., Braziel R.M., Rimsza L.M., Grogan T.M. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. 2004;351:2159–2169. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
