

Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress
- PMID: 30470534
- PMCID: PMC6859529
- DOI: 10.1016/j.redox.2018.11.005
Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress
Abstract
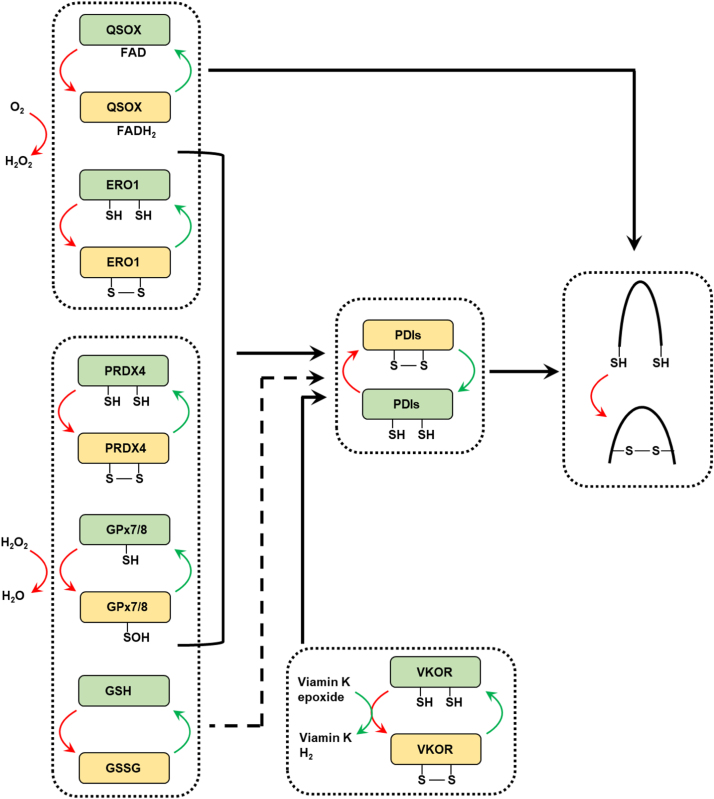
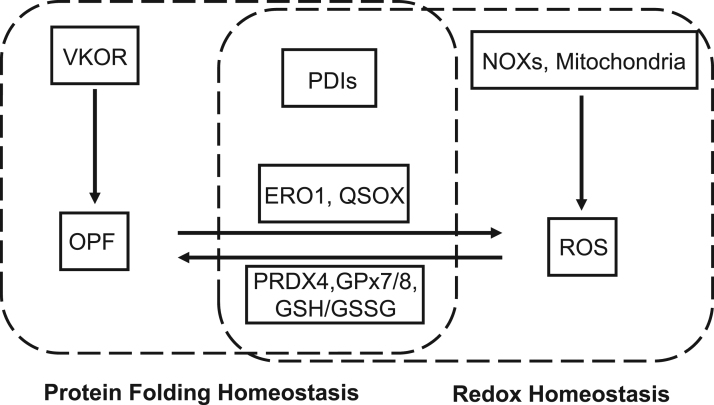
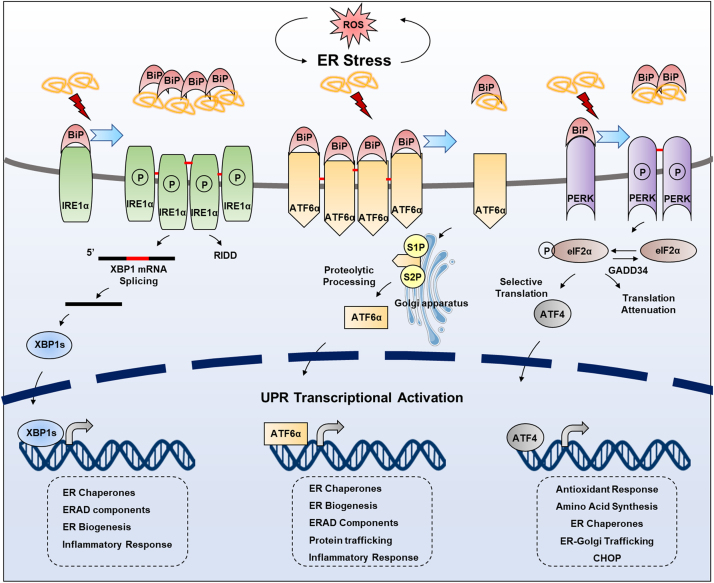
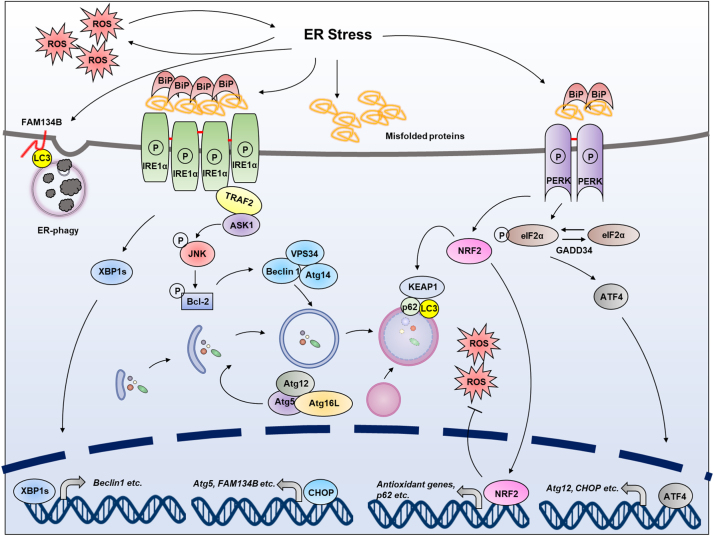
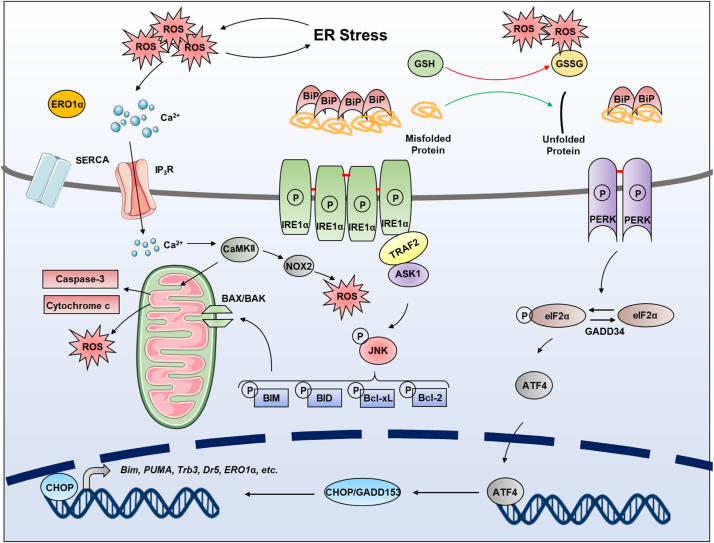
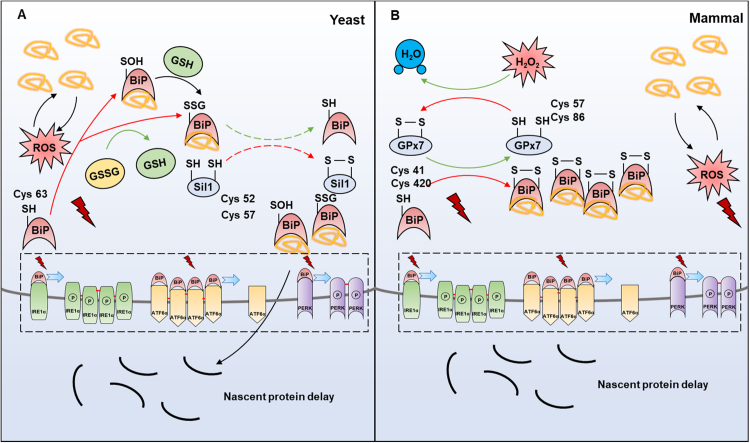
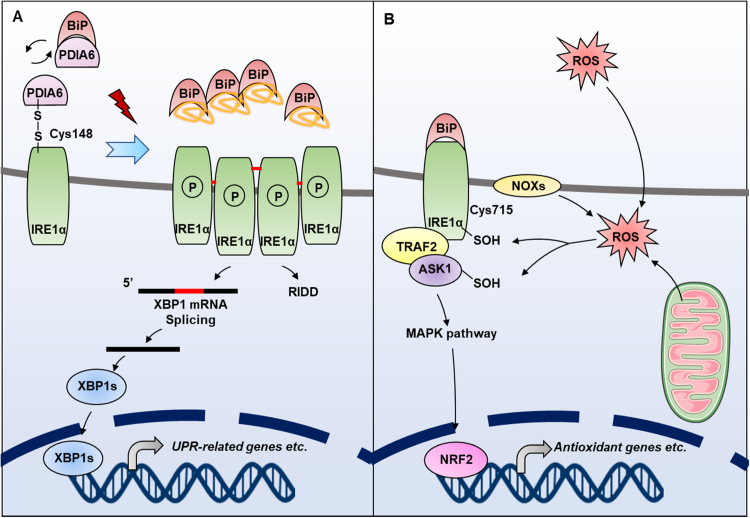
Endoplasmic reticulum (ER) is a dynamic organelle orchestrating the folding and post-translational maturation of almost all membrane proteins and most secreted proteins. These proteins synthesized in the ER, need to form disulfide bridge to acquire specific three-dimensional structures for function. The formation of disulfide bridge is mediated via protein disulfide isomerase (PDI) family and other oxidoreductases, which contribute to reactive oxygen species (ROS) generation and consumption in the ER. Therefore, redox regulation of ER is delicate and sensitive to perturbation. Deregulation in ER homeostasis, usually called ER stress, can provoke unfolded protein response (UPR) pathways with an aim to initially restore homeostasis by activating genes involved in protein folding and antioxidative machinery. Over time, however, activated UPR involves a variety of cellular signaling pathways which determine the state and fate of cell in large part (like autophagy, apoptosis, ferroptosis, inflammation, senescence, stemness, and cell cycle, etc.). This review will describe the regulation of UPR from the redox perspective in controlling the cell survival or death, emphasizing the redox modifications of UPR sensors/transducers in the ER.
Keywords: Cell fate; ER stress; Redox regulation; UPR.
Copyright © 2018 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Malhotra J.D., Kaufman R.J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007;9(12):2277–2293. - PubMed
-
- Enyedi B., Varnai P., Geiszt M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal. 2010;13(6):721–729. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
