

Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity
- PMID: 30470758
- PMCID: PMC6251914
- DOI: 10.1038/s41467-018-07384-7
Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity
Abstract
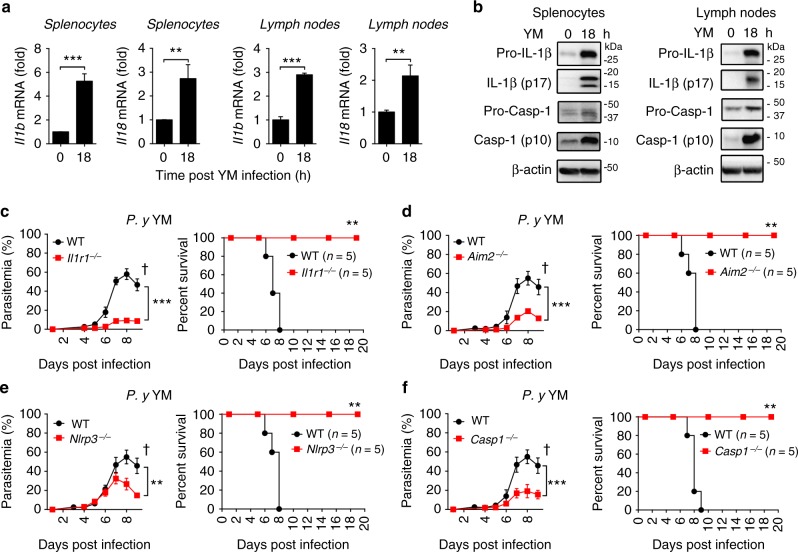
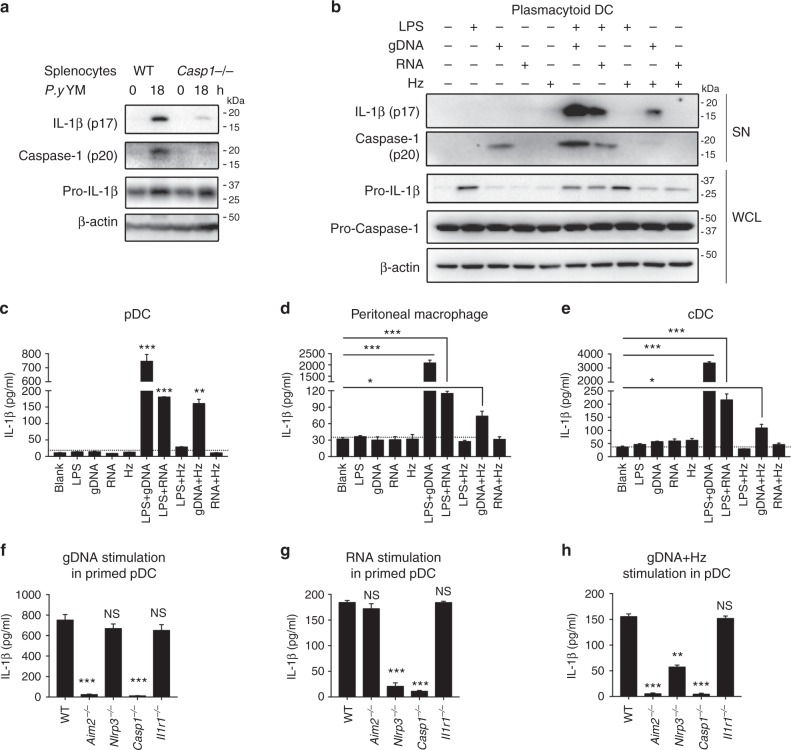
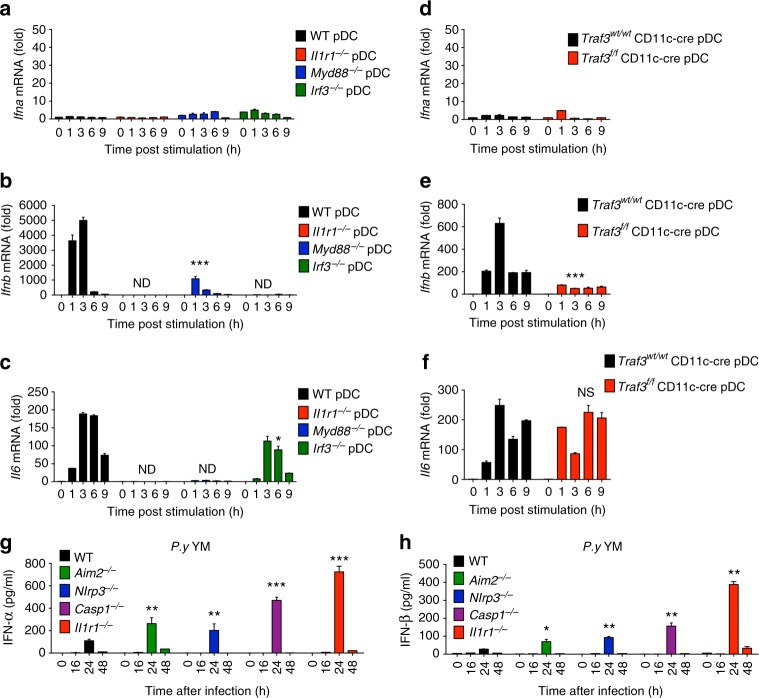
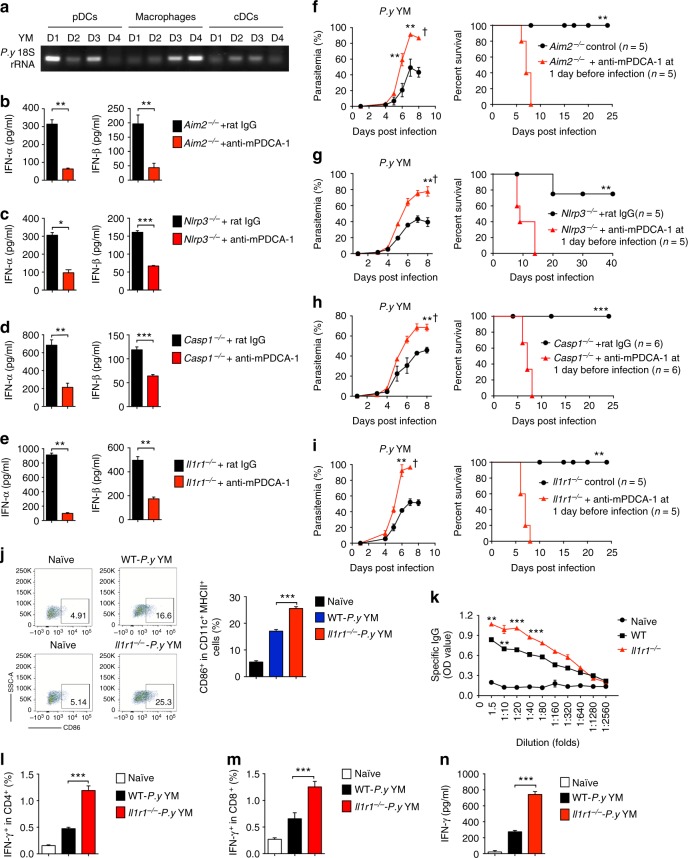
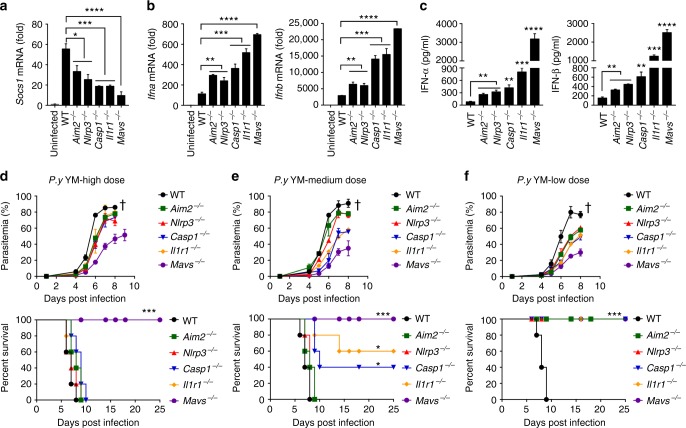
The inflammasome plays a critical role in inflammation and immune responses against pathogens. However, whether or how inflammasome activation regulates type I interferon (IFN-I) signaling in the context of malaria infection remain unknown. Here we show mice deficient in inflammasome sensors AIM2, NLRP3 or adaptor Caspase-1 produce high levels of IFN-I cytokines and are resistant to lethal Plasmodium yoelii YM infection. Inactivation of inflammasome signaling reduces interleukin (IL)-1β production, but increases IFN-I production. Mechanistically, we show inflammsome activation enhances IL-1β-mediated MyD88-TRAF3-IRF3 signaling and SOCS1 upregulation. However, SOCS1 inhibits MyD88-IRF7-mediated-IFN-I signaling and cytokine production in plasmacytoid dendritic cells. By contrast, ablation of inflammsome components reduces SOCS1 induction, and relieves its inhibition on MyD88-IRF7-dependent-IFN-I signaling, leading to high levels of IFN-α/β production and host survival. Our study identifies a previously unrecognized role of inflammasome activation in the negative regulation of IFN-I signaling pathways and provides potential targets for developing effective malaria vaccines.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
