

Antiangiogenic Gene Therapy in Cancer
- PMID: 30473624
- PMCID: PMC6247806
- DOI: 10.2174/1389202003351535
Antiangiogenic Gene Therapy in Cancer
Abstract
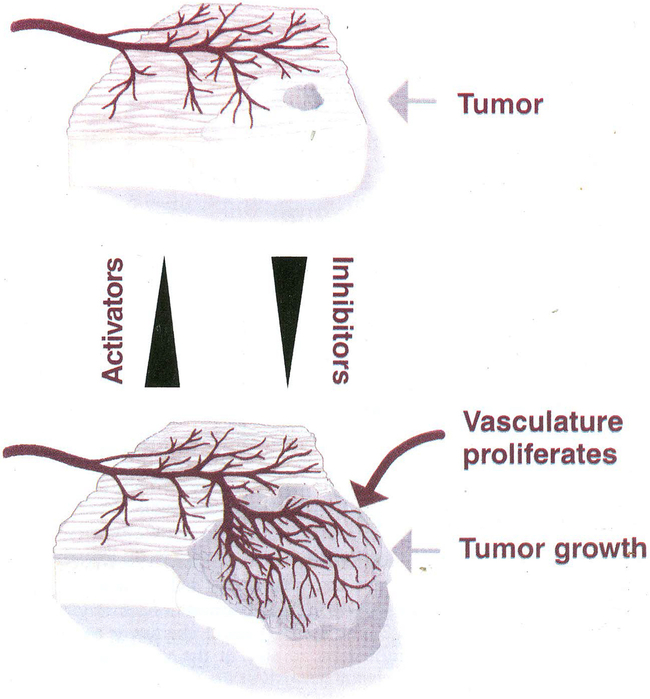
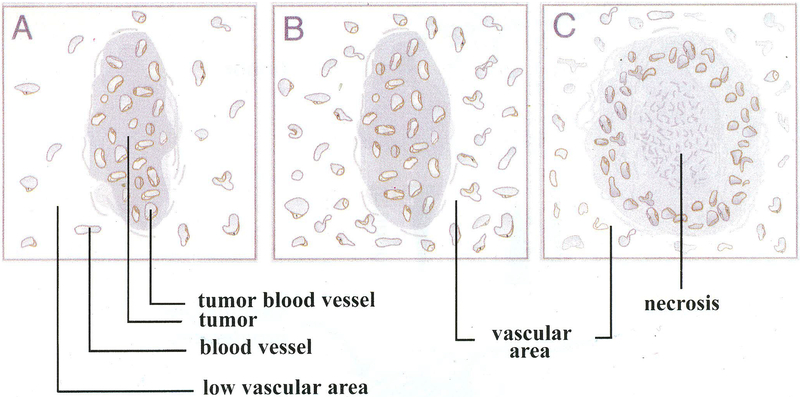
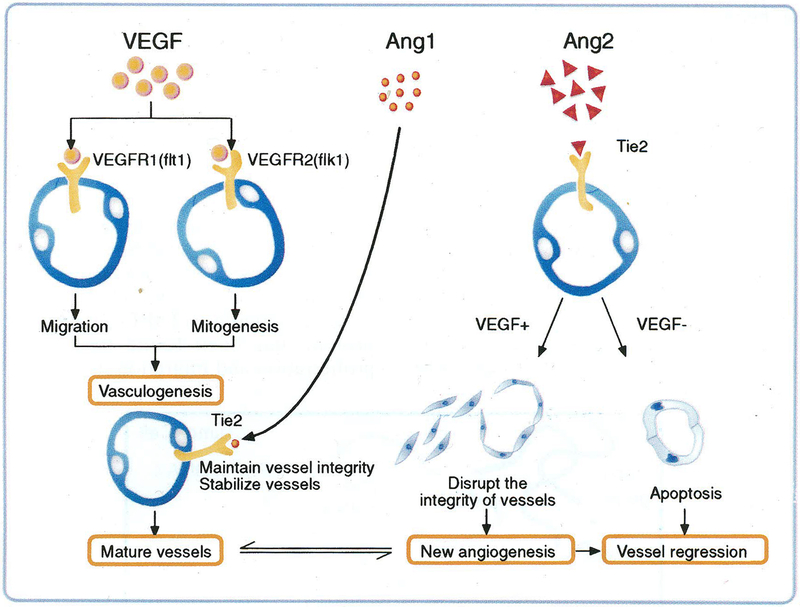
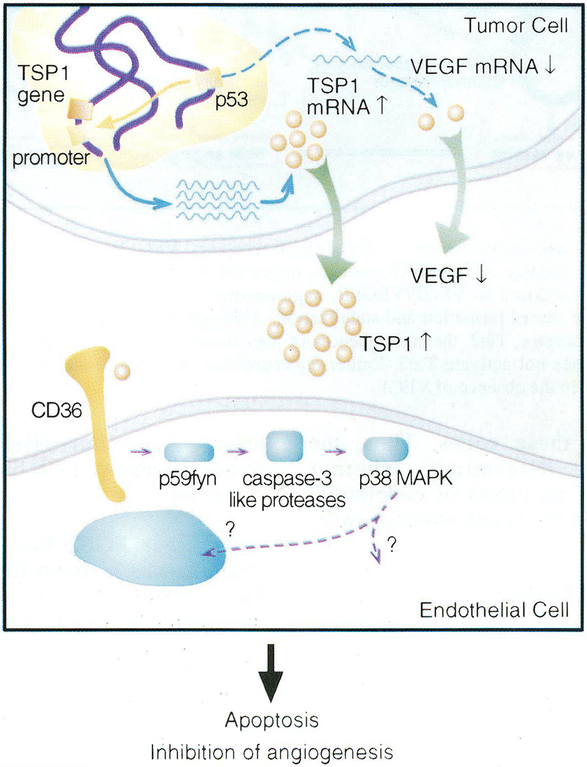
One of the most recent and exciting approaches in cancer gene therapy is the ability to target the developing blood supply of the tumor. An appealing feature of antiangiogenic gene therapy is that the tumor vasculature is a readily accessible target, particularly when the carrier and its gene are administered systemically. This is in contrast to several other gene therapy approaches in which the tumor vasculature represents a major obstacle to achieving high levels of transfection of the tumor cells. Several gene-based viral or non-viral therapies that target tumor angiogenesis have shown efficacy in pre-clinical models. Genes that encode antiangiogenic polypeptides such as angiostatin and endostatin have significantly inhibited tumor growth, inducing a microscopic dormant state. The products of these genes are thought to act extracellularly to inhibit angiogenesis. An alternative approach that investigators have used successfully in tumor-bearing mice is to target angiogenic growth factors or their receptors that are essential for tumor growth. Levels of angiogenic factors such as vascular endothelial growth factor (VEGF) have been reduced by either antisense methods or the use of genes encoding truncated angiogenic decoy receptors. Despite these promising findings of tumor reduction with antiangiogenic gene therapy, advances in the viral and/or non-viral delivery systems are essential for this therapy to have clinical utility. In this review, we will discuss the mechanisms of angiogenesis/antiangiogenesis, and the current status and future directions of antiangiogenic gene therapy.
Figures
Similar articles
-
Targeting tumor angiogenesis with gene therapy.Mol Genet Metab. 2001 Sep-Oct;74(1-2):120-7. doi: 10.1006/mgme.2001.3223. Mol Genet Metab. 2001. PMID: 11592809 Review.
-
Adeno-associated virus 2-mediated antiangiogenic cancer gene therapy: long-term efficacy of a vector encoding angiostatin and endostatin over vectors encoding a single factor.Cancer Res. 2004 Mar 1;64(5):1781-7. doi: 10.1158/0008-5472.can-03-1786. Cancer Res. 2004. PMID: 14996740
-
Adeno-associated virus-mediated antiangiogenic gene therapy with thrombospondin-1 type 1 repeats and endostatin.Clin Cancer Res. 2007 Jul 1;13(13):3968-76. doi: 10.1158/1078-0432.CCR-07-0245. Clin Cancer Res. 2007. PMID: 17606731
-
Combination antiangiogenic therapy and radiation in head and neck cancers.Oral Oncol. 2014 Jan;50(1):19-26. doi: 10.1016/j.oraloncology.2013.10.003. Epub 2013 Oct 23. Oral Oncol. 2014. PMID: 24269532 Review.
-
Antiangiogenic gene therapy for hepatocellular carcinoma using angiostatin gene.Hepatology. 2003 Mar;37(3):696-704. doi: 10.1053/jhep.2003.50077. Hepatology. 2003. PMID: 12601367
References
-
- Folkman J Tumor angiogenesis: Therapeutic implications, N. Eng. J. Med 333: 1757–1763, 1971. - PubMed
-
- Folkman J The vasculization of tumors, Sci. Am 234: 58–73, 1976. - PubMed
-
- Kerbel RS Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents, Bioessays. 13: 31–36, 1991. - PubMed
-
- Boehm T, Folkman J, Browder T, and O’Reilly MS Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance [see comments], Nature. 390: 404–407, 1997. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources