

Deep repeat resolution-the assembly of the Drosophila Histone Complex
- PMID: 30476267
- PMCID: PMC6380962
- DOI: 10.1093/nar/gky1194
Deep repeat resolution-the assembly of the Drosophila Histone Complex
Abstract
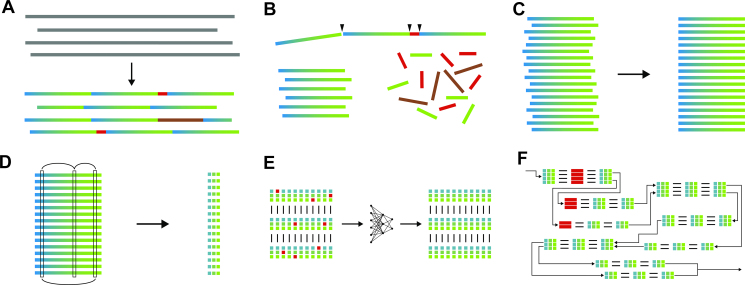
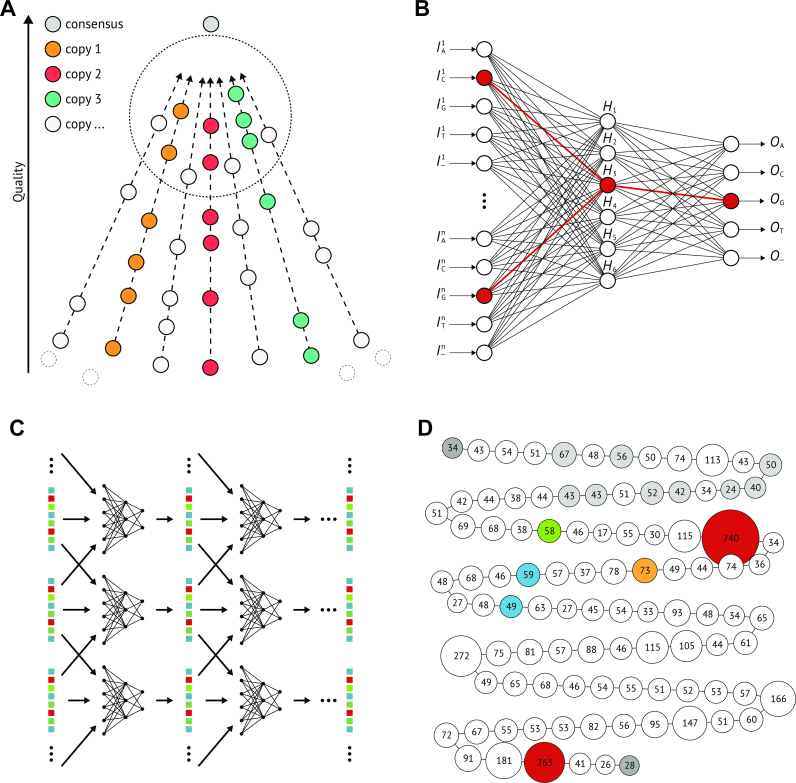
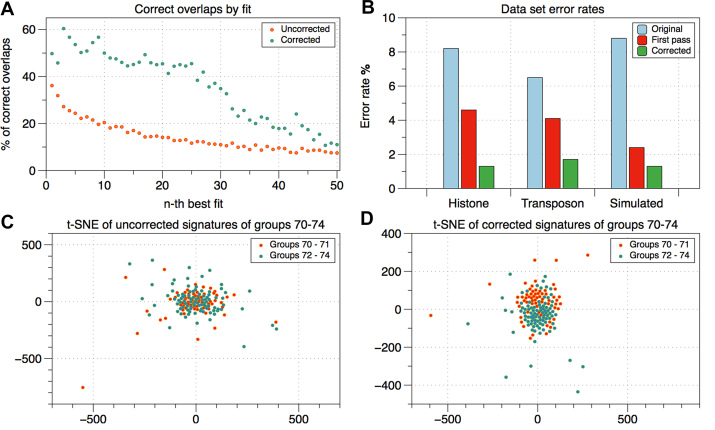
Though the advent of long-read sequencing technologies has led to a leap in contiguity of de novo genome assemblies, current reference genomes of higher organisms still do not provide unbroken sequences of complete chromosomes. Despite reads in excess of 30 000 base pairs, there are still repetitive structures that cannot be resolved by current state-of-the-art assemblers. The most challenging of these structures are tandemly arrayed repeats, which occur in the genomes of all eukaryotes. Untangling tandem repeat clusters is exceptionally difficult, since the rare differences between repeat copies are obscured by the high error rate of long reads. Solving this problem would constitute a major step towards computing fully assembled genomes. Here, we demonstrate by example of the Drosophila Histone Complex that via machine learning algorithms, it is possible to exploit the underlying distinguishing patterns of single nucleotide variants of repeats from very noisy data to resolve a large and highly conserved repeat cluster. The ideas explored in this paper are a first step towards the automated assembly of complex repeat structures and promise to be applicable to a wide range of eukaryotic genomes.
© The Author(s) 2018. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Morgan T.H. An attempt to analyze the constitution of the chromosomes on the basis of sex-limited inheritance in drosophila. J. Exp. Zool. Part A. 1911; 11:365–413.
-
- Myers E.W., Sutton G., Delcher A., Dew I., Fasulo D., Flanigan M., Kravitz S., Mobarry C., Reinert K., Remington K. et al. .. A whole-genome assembly of drosophila. Science. 2000; 287:2196–2204. - PubMed
-
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. - PubMed
-
- Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A. et al. .. The sequence of the human genome. Science. 2001; 291:1304–1351. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
