

Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History
- PMID: 30477216
- PMCID: PMC6306719
- DOI: 10.3390/jcm7120476
Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History
Abstract
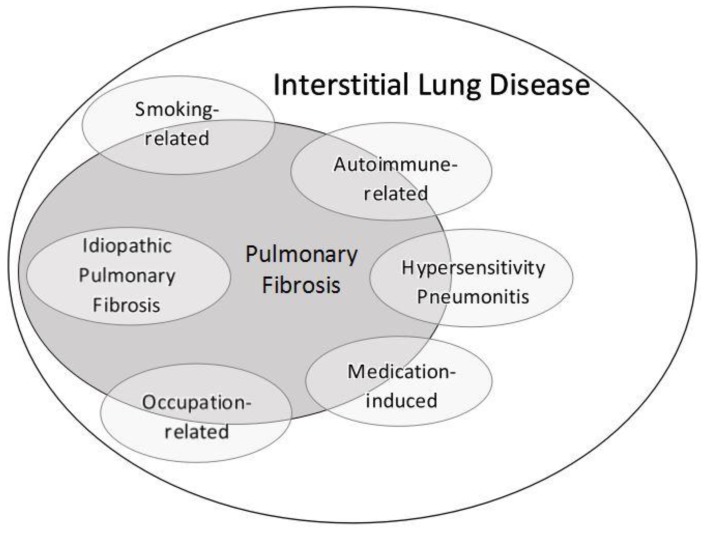
Interstitial lung disease (ILD) and pulmonary fibrosis comprise a wide array of inflammatory and fibrotic lung diseases which are often confusing to general medicine and pulmonary physicians alike. In addition to the myriad of clinical and radiologic nomenclature used in ILD, histopathologic descriptors may be particularly confusing, and are often extrapolated to radiologic imaging patterns which may further add to the confusion. We propose that rather than focusing on precise histologic findings, focus should be on identifying an accurate etiology of ILD through a comprehensive and detailed medical history. Histopathologic patterns from lung biopsy should not be dismissed, but are often nonspecific, and overall treatment strategy and prognosis are likely to be determined more by the specific etiology of ILD rather than any particular histologic pattern. In this review, we outline a practical approach to common ILDs, highlight important aspects in obtaining an exposure history, clarify terminology and nomenclature, and discuss six common subgroups of ILD likely to be encountered by general medicine physicians in the inpatient or outpatient setting: Smoking-related, hypersensitivity pneumonitis, connective tissue disease-related, occupation-related, medication-induced, and idiopathic pulmonary fibrosis. Accurate diagnosis of these forms of ILD does require supplementing the medical history with results of the physical examination, autoimmune serologic testing, and chest radiographic imaging, but the importance of a comprehensive environmental, avocational, occupational, and medication-use history cannot be overstated and is likely the single most important factor responsible for achieving the best possible outcomes for patients.
Keywords: autoimmune; hypersensitivity; idiopathic pulmonary fibrosis; occupation; smoking.
Conflict of interest statement
The authors declare no conflict of interest related to this manuscript.
Figures
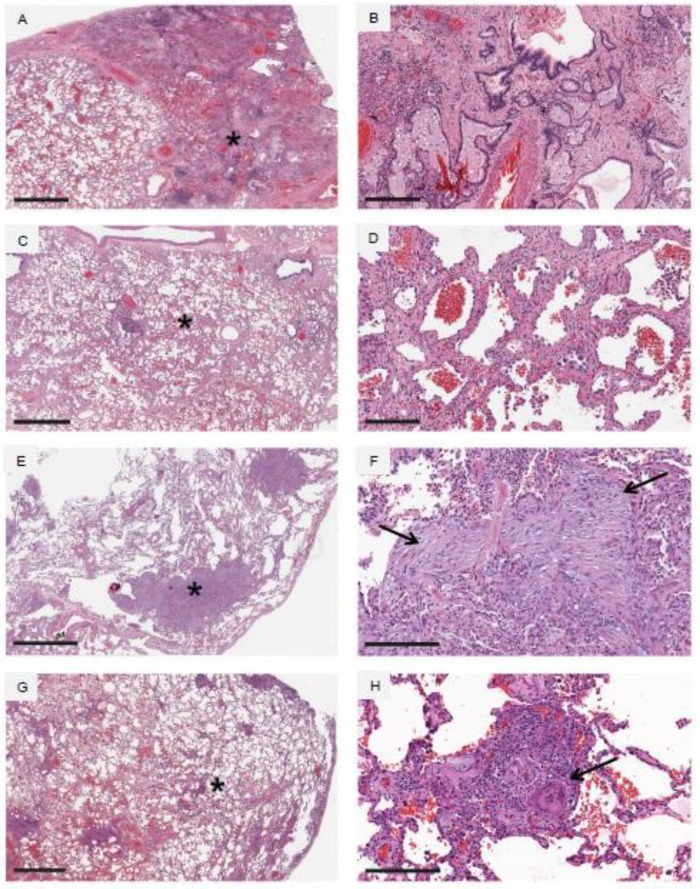
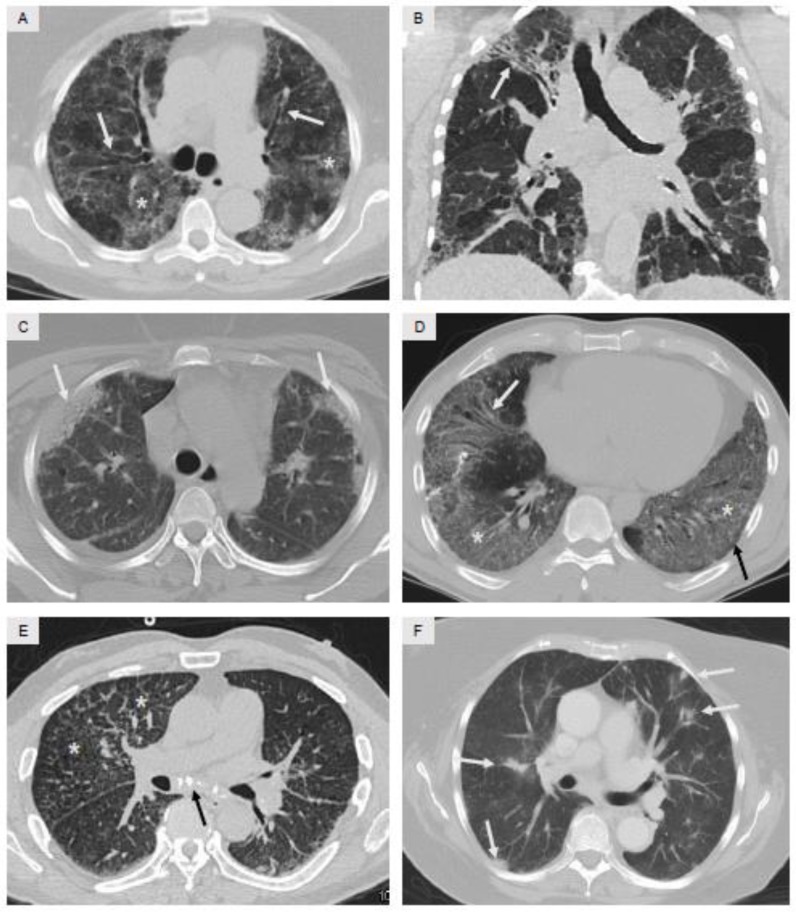
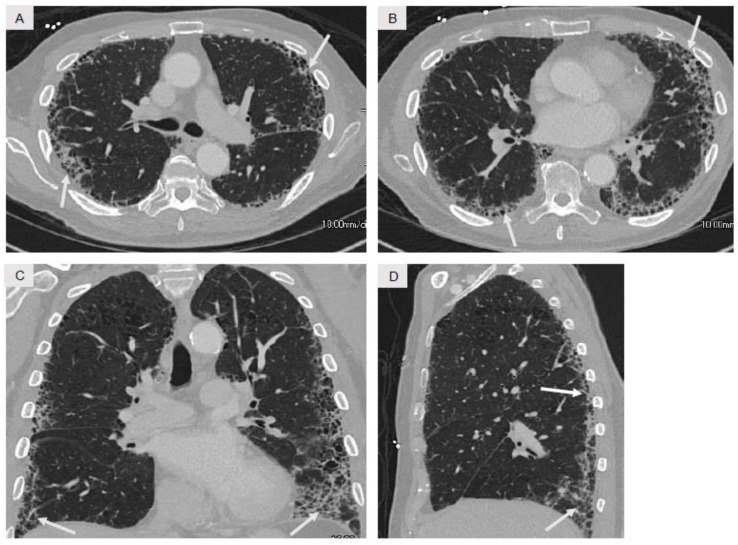
References
-
- American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am. J. Respir. Crit. Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. - DOI - PubMed
-
- Travis W.D., Costabel U., Hansell D.M., King T.E.J., Lynch D.A., Nicholson A.G., Ryerson C.J., Ryu J.H., Selman M., Wells A.U., et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. - DOI - PMC - PubMed
-
- Bradley B., Branley H.M., Egan J.J., Greaves M.S., Hansell D.M., Harrison N.K., Hirani N., Hubbard R., Lake F., Millar A.B., et al. Interstitial lung disease guideline: The British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63(Suppl. 5):v1–v58. - PubMed
-
- Raghu G., Collard H.R., Egan J.J., Martinez F.J., Behr J., Brown K.K., Colby T.V., Cordier J.-F., Flaherty K.R., Lasky J.A., et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. - DOI - PMC - PubMed
-
- Raghu G., Rochwerg B., Zhang Y., Garcia C.A.C., Azuma A., Behr J., Brozek J.L., Collard H.R., Cunningham W., Homma S., et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015;192:e3–e19. doi: 10.1164/rccm.201506-1063ST. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
