

[Clinical and genetic features of Kallmann syndrome: an analysis of 5 cases]
- PMID: 30477624
- PMCID: PMC7389016
- DOI: 10.7499/j.issn.1008-8830.2018.11.009
[Clinical and genetic features of Kallmann syndrome: an analysis of 5 cases]
Abstract
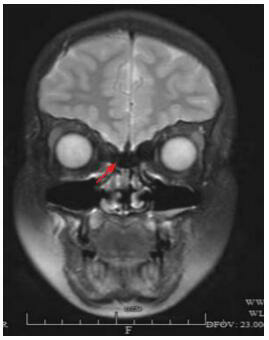
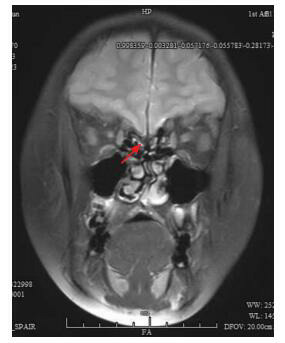
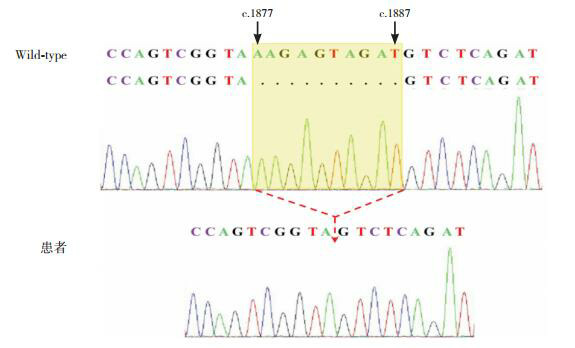
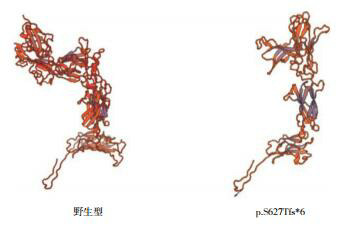
Kallmann syndrome (KS) is a rare pediatric disease with major manifestations of olfactory dysfunction and hypogonadotropic hypogonadism. Five children (4 boys and 1 girl) with KS reported in this article were aged between 6 months and 19 years at the time when they attended the hospital. All the children had the clinical manifestation of hypogonadotropic hypogonadism; in addition, three children had olfactory dysfunction (two were found to have olfactory bulb dysplasia on magnetic resonance imaging), one had cleft lip and palate, and one had micropenis and cryptorchidism with right renal agenesis during infancy. All the five children had normal karyotype and their parents had normal clinical phenotypes. The uncle of one child had underdeveloped secondary sexual characteristics and olfactory disorder since childhood. High-throughput sequencing found two known heterozygous missense mutations in the FGFR1 gene, i.e., c.1097C>T(p.P366L) and c.809G>C(p.G270A), in two children. One child had a novel frameshift mutation, c.1877_1887/p.S627Tfs*6, in the KAL1 gene; this deletion mutation caused a frameshift in base sequence and produced truncated proteins, which led to a significant change in protein structure, and thus it was highly pathogenic. It is concluded that KS has great clinical and genetic heterogeneity and can be accompanied by incomplete dominant inheritance and that gene detection helps with the diagnosis of this disease.
Kallmann综合征(KS)是一类罕见的儿科疾病,主要表现为嗅觉功能缺陷和低促性腺激素性腺功能减退。本文报道的5例KS患者就诊年龄为6月龄至19岁,男4例、女1例,均表现为低促性腺激素性腺功能减退,其中3例伴嗅觉功能缺陷(2例MRI发现嗅球发育不良),1例伴唇腭裂,1例婴儿期表现为小阴茎、隐睾、右肾缺如。5例患者染色体核型均正常,其父母临床表型均正常,1例的伯父自幼有第二性征发育不全伴嗅觉障碍。高通量测序发现2例存在已报道的FGFR1基因杂合错义突变,分别是c.1097C > T(p.P366L)、c.809G > C(p.G270A);1例存在KAL1基因的新发移码突变:c.1877_1887/p.S627Tfs*6,该缺失突变碱基序列移码并产生截短蛋白,蛋白结构明显改变,具有高度致病性。KS临床和遗传异质性大,可伴不完全显性遗传,基因检测有助于诊断。
Figures
Similar articles
-
[Children with idiopathic hypogonadotropic hypogonadism: clinical data analysis and mutations analysis of KAL1 and FGFR1 gene].Zhonghua Er Ke Za Zhi. 2014 Dec;52(12):942-7. Zhonghua Er Ke Za Zhi. 2014. PMID: 25619354 Chinese.
-
Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients.J Clin Endocrinol Metab. 2004 Mar;89(3):1079-88. doi: 10.1210/jc.2003-030476. J Clin Endocrinol Metab. 2004. PMID: 15001591
-
Mutation analyses in pedigrees and sporadic cases of ethnic Han Chinese Kallmann syndrome patients.Exp Biol Med (Maywood). 2015 Nov;240(11):1480-9. doi: 10.1177/1535370215587531. Epub 2015 Jun 1. Exp Biol Med (Maywood). 2015. PMID: 26031747 Free PMC article.
-
Reversible Kallmann syndrome: report of the first case with a KAL1 mutation and literature review.Eur J Endocrinol. 2007 Mar;156(3):285-90. doi: 10.1530/eje.1.02342. Eur J Endocrinol. 2007. PMID: 17322486 Review.
-
[Molecular genetics of Kallmann syndrome: an update].Zhonghua Nan Ke Xue. 2011 Apr;17(4):361-5. Zhonghua Nan Ke Xue. 2011. PMID: 21548217 Review. Chinese.
Cited by
-
Kallmann Syndrome Due to Heterozygous Mutation in SOX10 Coexisting With Waardenburg Syndrome Type II: Case Report and Review of Literature.Front Endocrinol (Lausanne). 2021 Feb 1;11:592831. doi: 10.3389/fendo.2020.592831. eCollection 2020. Front Endocrinol (Lausanne). 2021. PMID: 33597923 Free PMC article. Review.
-
Clinical manifestations and spermatogenesis outcomes in Chinese patients with congenital hypogonadotropic hypogonadism caused by inherited or de novo FGFR1 mutations.Asian J Androl. 2024 Jul 1;26(4):426-432. doi: 10.4103/aja202366. Epub 2024 Jan 9. Asian J Androl. 2024. PMID: 38227553 Free PMC article.
References
-
- Dodé C, Fouveaut C, Mortier G, et al. Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis. http://cn.bing.com/academic/profile?id=4ccc060d1f5a627df03bf8d908a89751&... Hum Mutat. 2007;28(1):97–98. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous