

VDAC2 enables BAX to mediate apoptosis and limit tumor development
- PMID: 30478310
- PMCID: PMC6255874
- DOI: 10.1038/s41467-018-07309-4
VDAC2 enables BAX to mediate apoptosis and limit tumor development
Abstract
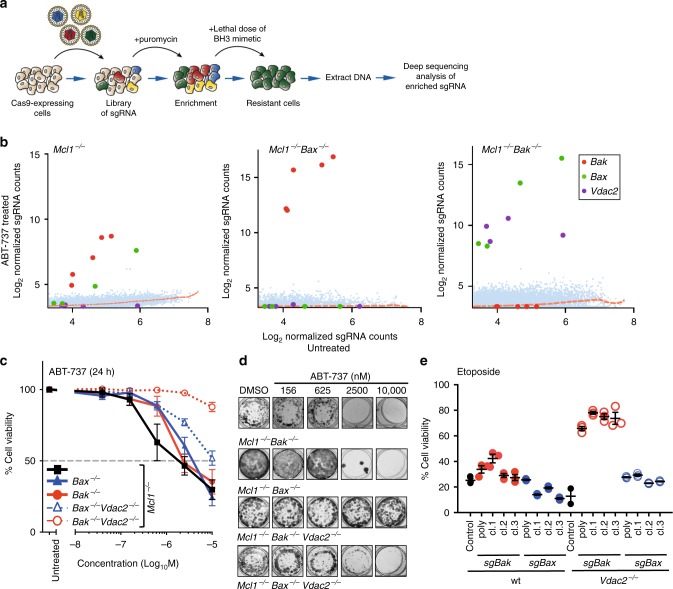
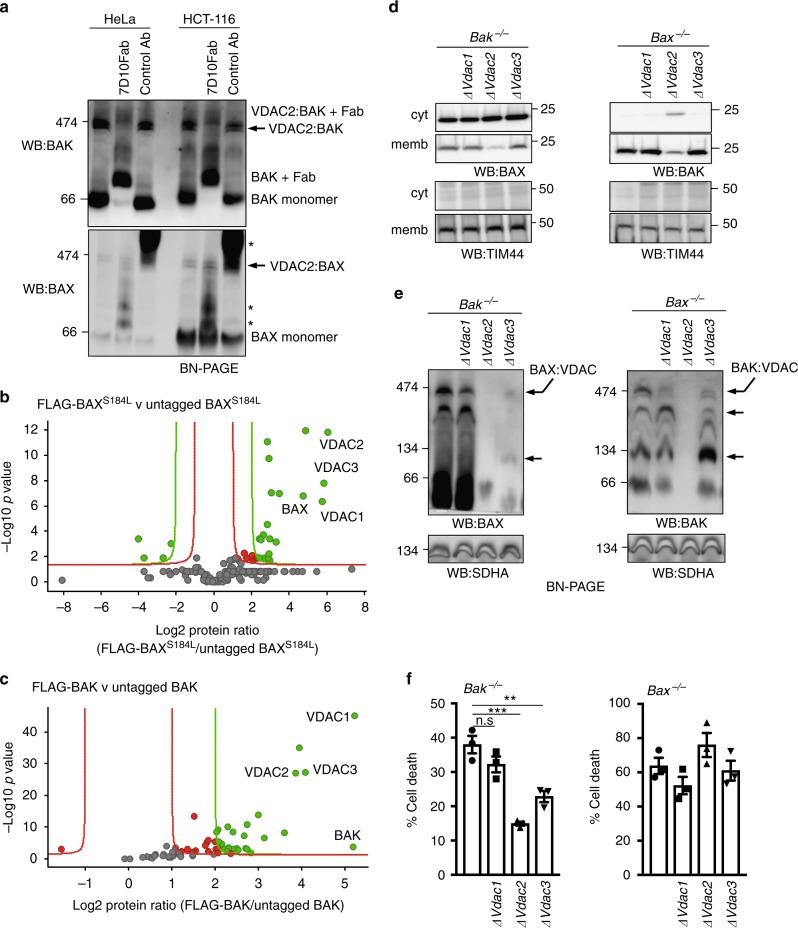
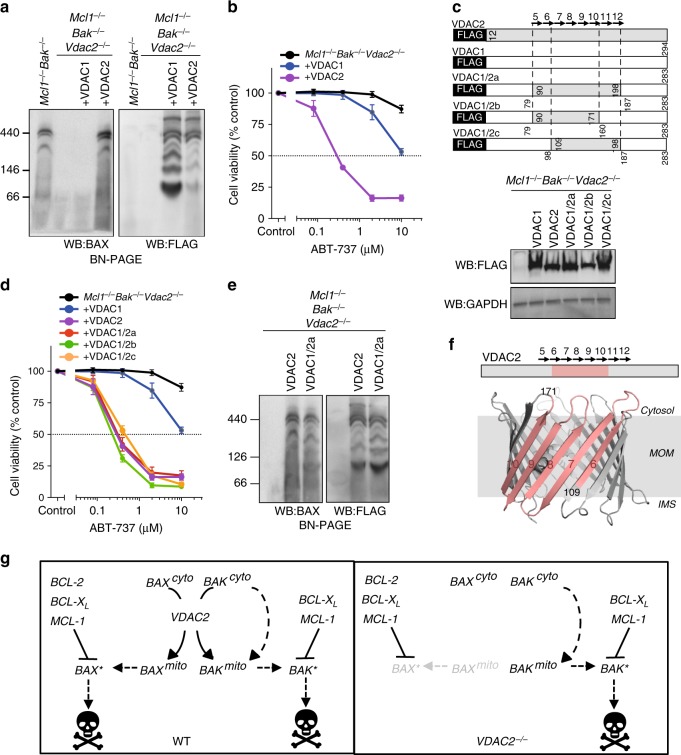
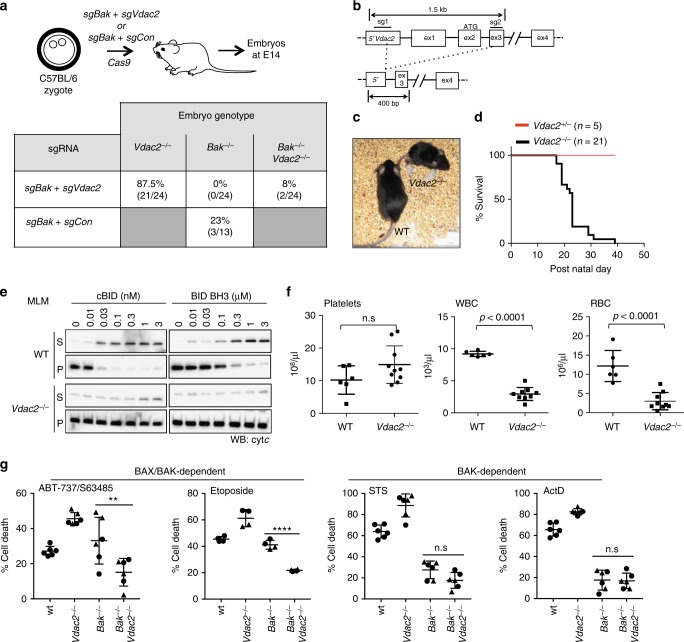
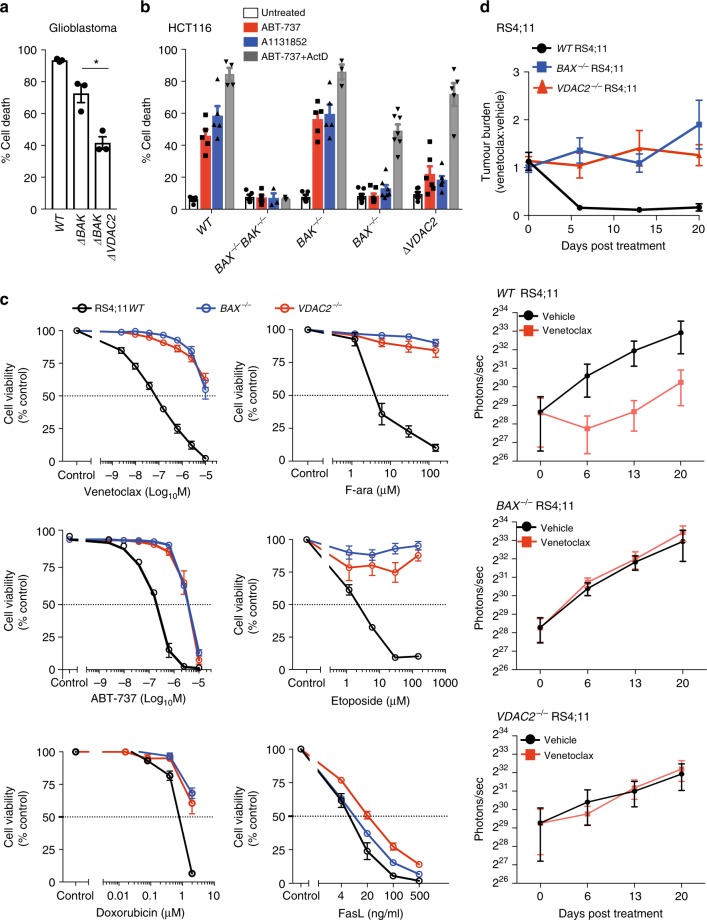
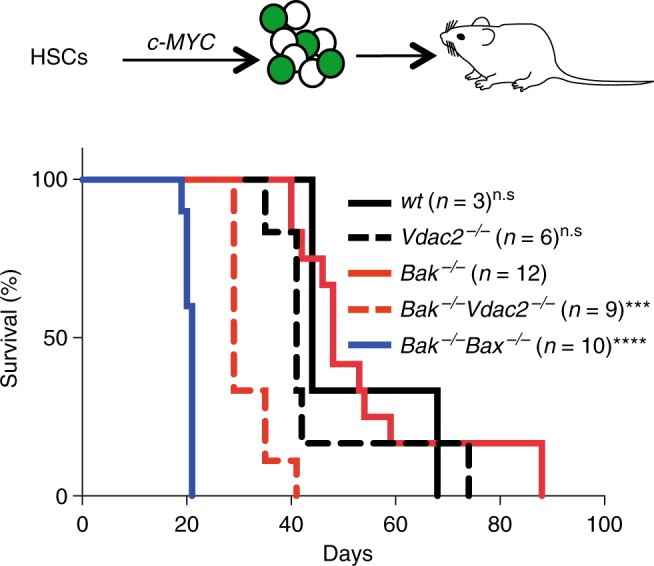
Intrinsic apoptosis is critical to prevent tumor formation and is engaged by many anti-cancer agents to eliminate tumor cells. BAX and BAK, the two essential mediators of apoptosis, are thought to be regulated through similar mechanisms and act redundantly to drive apoptotic cell death. From an unbiased genome-wide CRISPR/Cas9 screen, we identified VDAC2 (voltage-dependent anion channel 2) as important for BAX, but not BAK, to function. Genetic deletion of VDAC2 abrogated the association of BAX and BAK with mitochondrial complexes containing VDAC1, VDAC2, and VDAC3, but only inhibited BAX apoptotic function. Deleting VDAC2 phenocopied the loss of BAX in impairing both the killing of tumor cells by anti-cancer agents and the ability to suppress tumor formation. Together, our studies show that efficient BAX-mediated apoptosis depends on VDAC2, and reveal a striking difference in how BAX and BAK are functionally impacted by their interactions with VDAC2.
Conflict of interest statement
M.Fv.D., D.H.D.G., C.C., C.M.H., G.L.K., I.K.L.T., L.F.D., A.W., J.J.S., A.J.K., M.J.H., K.B., A.L.S., B.R., P.B., R.M.K., D.C.S.H., and G.D. are employees of the Walter and Eliza Hall Institute of Medical Research, which receives milestone payments for venetoclax. All other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
