

Quantifying post-transcriptional regulation in the development of Drosophila melanogaster
- PMID: 30478415
- PMCID: PMC6255845
- DOI: 10.1038/s41467-018-07455-9
Quantifying post-transcriptional regulation in the development of Drosophila melanogaster
Abstract
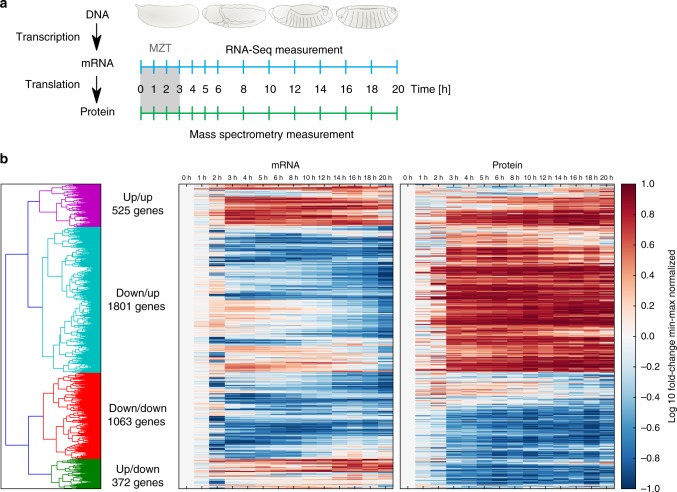
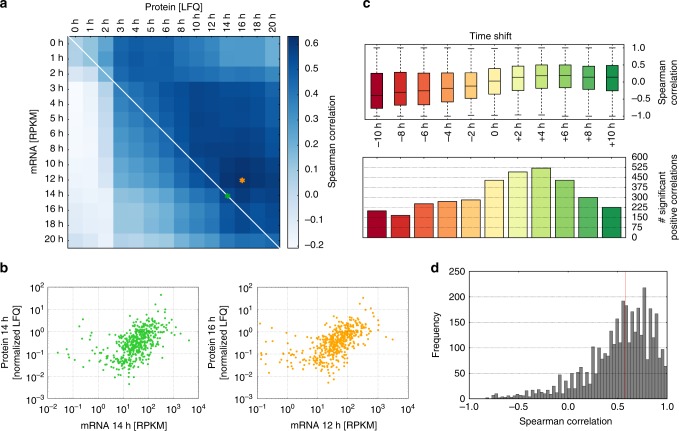
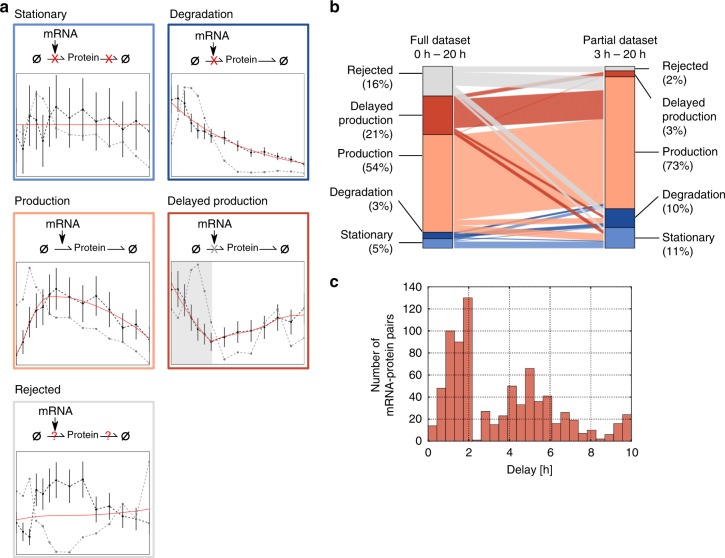
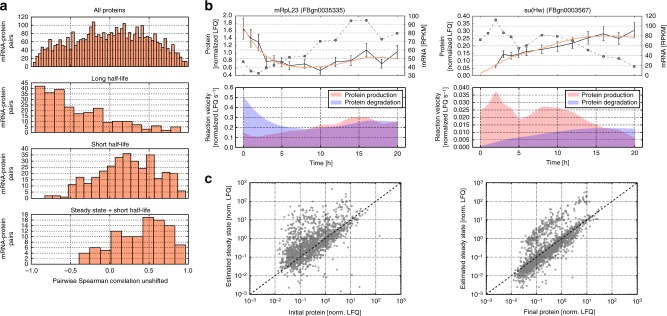
Even though proteins are produced from mRNA, the correlation between mRNA levels and protein abundances is moderate in most studies, occasionally attributed to complex post-transcriptional regulation. To address this, we generate a paired transcriptome/proteome time course dataset with 14 time points during Drosophila embryogenesis. Despite a limited mRNA-protein correlation (ρ = 0.54), mathematical models describing protein translation and degradation explain 84% of protein time-courses based on the measured mRNA dynamics without assuming complex post transcriptional regulation, and allow for classification of most proteins into four distinct regulatory scenarios. By performing an in-depth characterization of the putatively post-transcriptionally regulated genes, we postulate that the RNA-binding protein Hrb98DE is involved in post-transcriptional control of sugar metabolism in early embryogenesis and partially validate this hypothesis using Hrb98DE knockdown. In summary, we present a systems biology framework for the identification of post-transcriptional gene regulation from large-scale, time-resolved transcriptome and proteome data.
Conflict of interest statement
S.S. is currently employed at Boehringer Ingelheim International GmbH. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
