

DNA methylation dynamics during embryonic development and postnatal maturation of the mouse auditory sensory epithelium
- PMID: 30478432
- PMCID: PMC6255903
- DOI: 10.1038/s41598-018-35587-x
DNA methylation dynamics during embryonic development and postnatal maturation of the mouse auditory sensory epithelium
Abstract
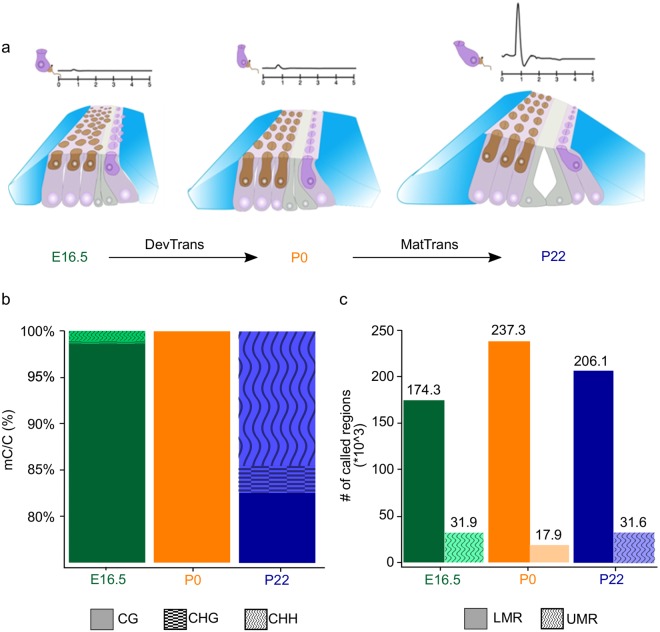
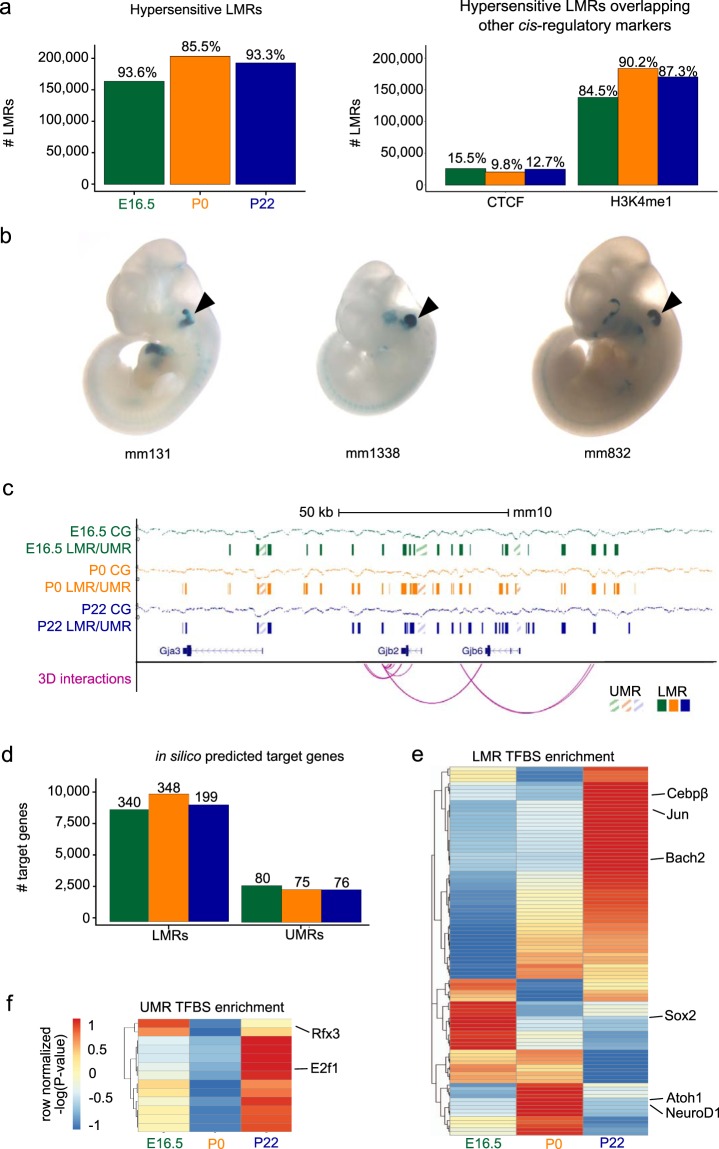
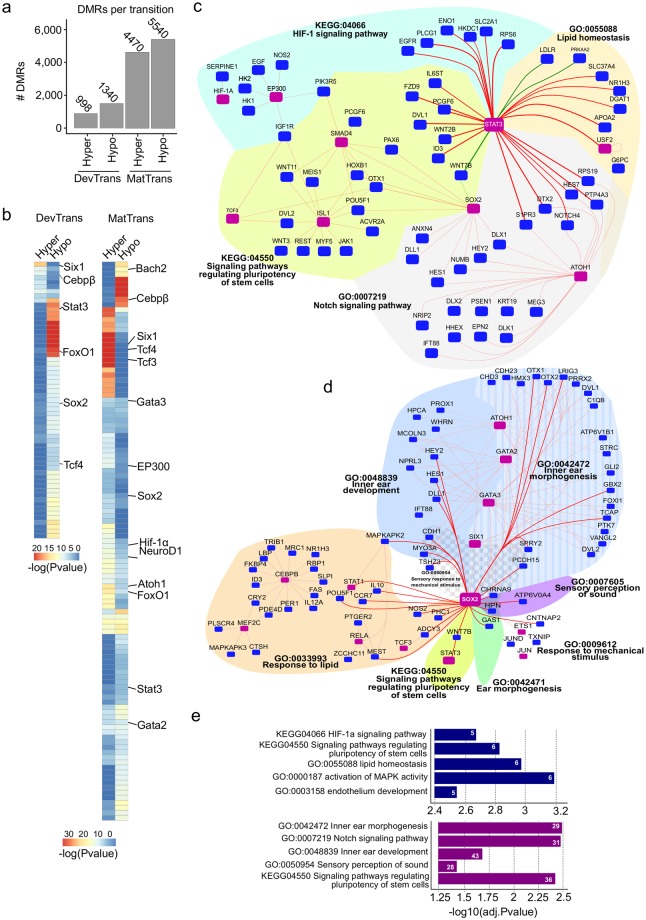
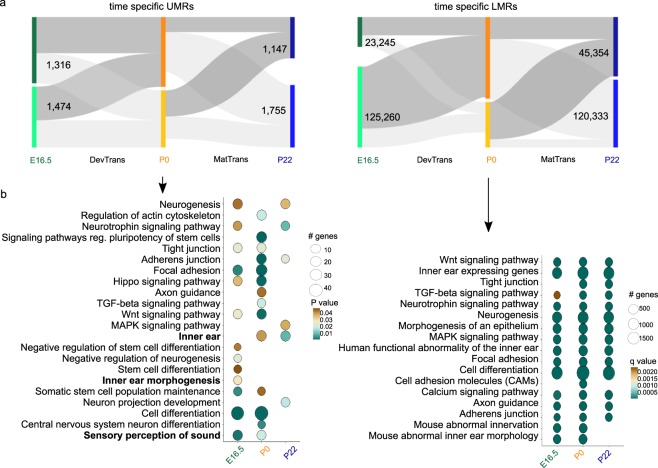
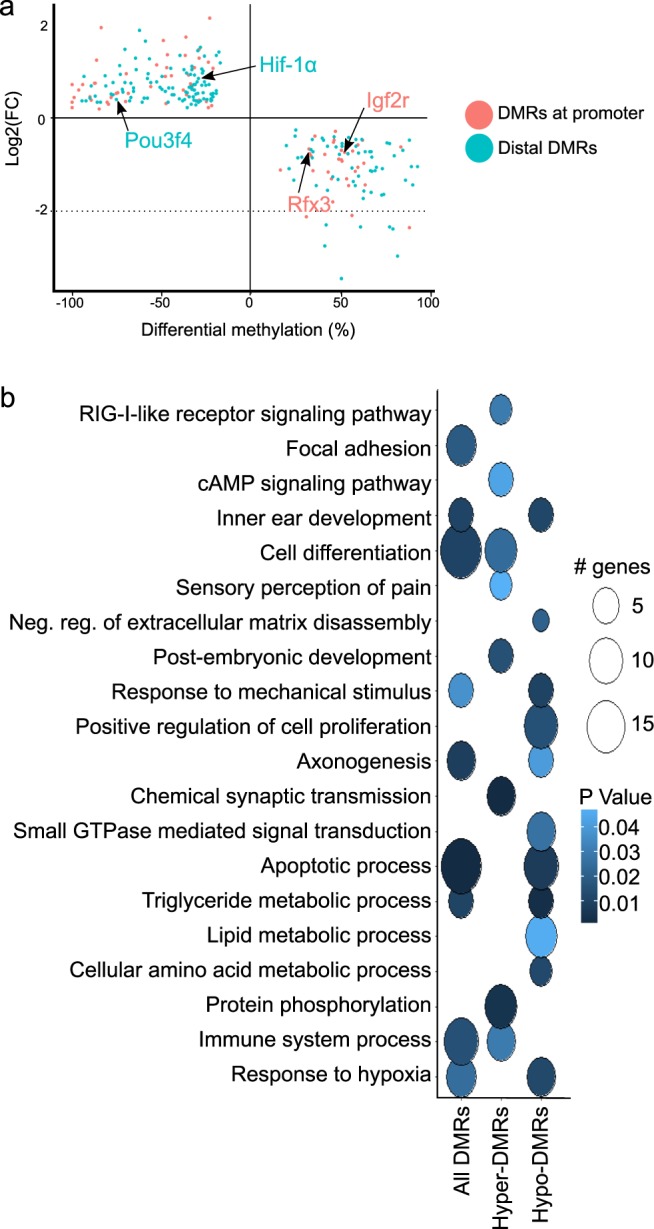
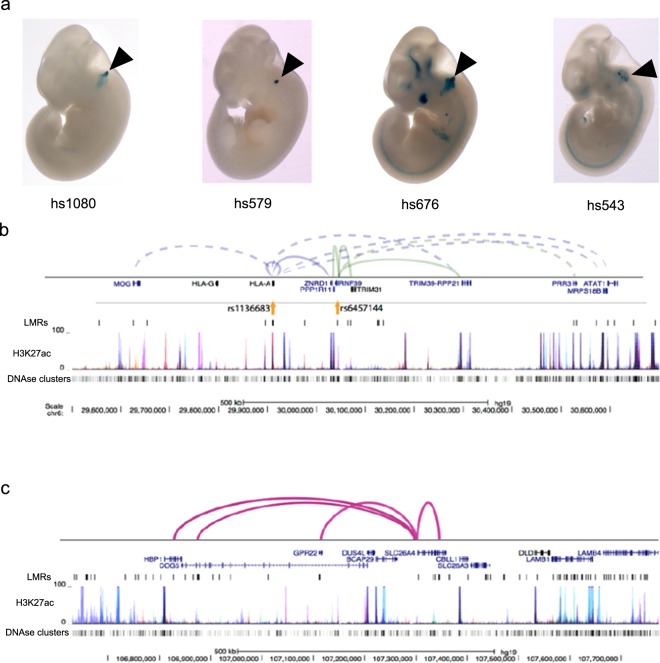
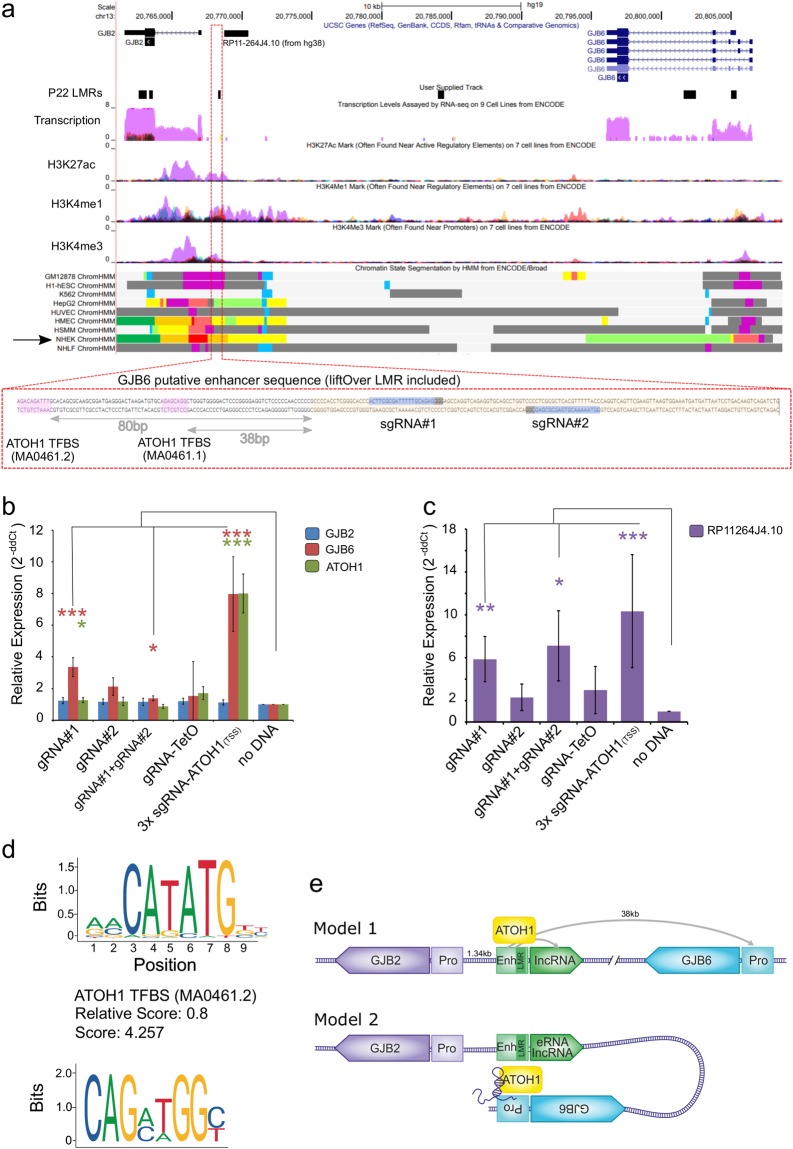
The inner ear is a complex structure responsible for hearing and balance, and organ pathology is associated with deafness and balance disorders. To evaluate the role of epigenomic dynamics, we performed whole genome bisulfite sequencing at key time points during the development and maturation of the mouse inner ear sensory epithelium (SE). Our single-nucleotide resolution maps revealed variations in both general characteristics and dynamics of DNA methylation over time. This allowed us to predict the location of non-coding regulatory regions and to identify several novel candidate regulatory factors, such as Bach2, that connect stage-specific regulatory elements to molecular features that drive the development and maturation of the SE. Constructing in silico regulatory networks around sites of differential methylation enabled us to link key inner ear regulators, such as Atoh1 and Stat3, to pathways responsible for cell lineage determination and maturation, such as the Notch pathway. We also discovered that a putative enhancer, defined as a low methylated region (LMR), can upregulate the GJB6 gene and a neighboring non-coding RNA. The study of inner ear SE methylomes revealed novel regulatory regions in the hearing organ, which may improve diagnostic capabilities, and has the potential to guide the development of therapeutics for hearing loss by providing multiple intervention points for manipulation of the auditory system.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
