

Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions
- PMID: 30478443
- PMCID: PMC6520989
- DOI: 10.1038/s41588-018-0274-x
Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions
Abstract
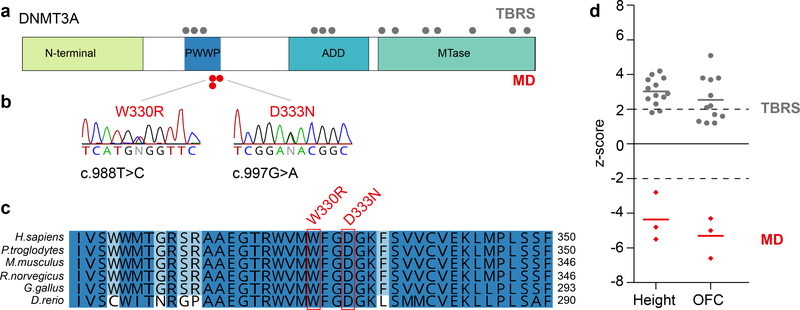
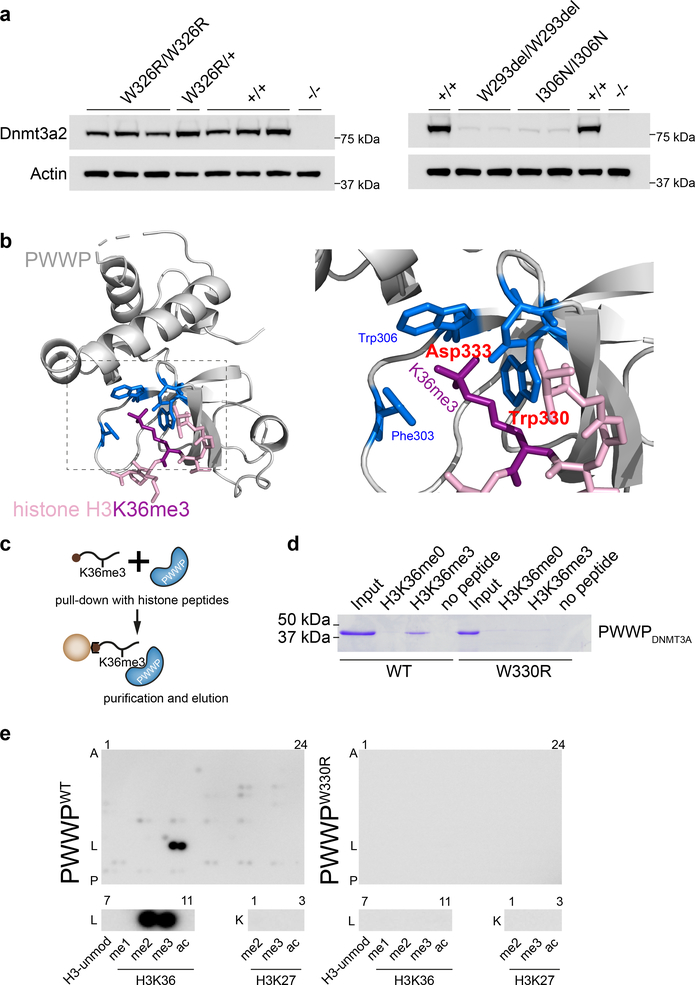
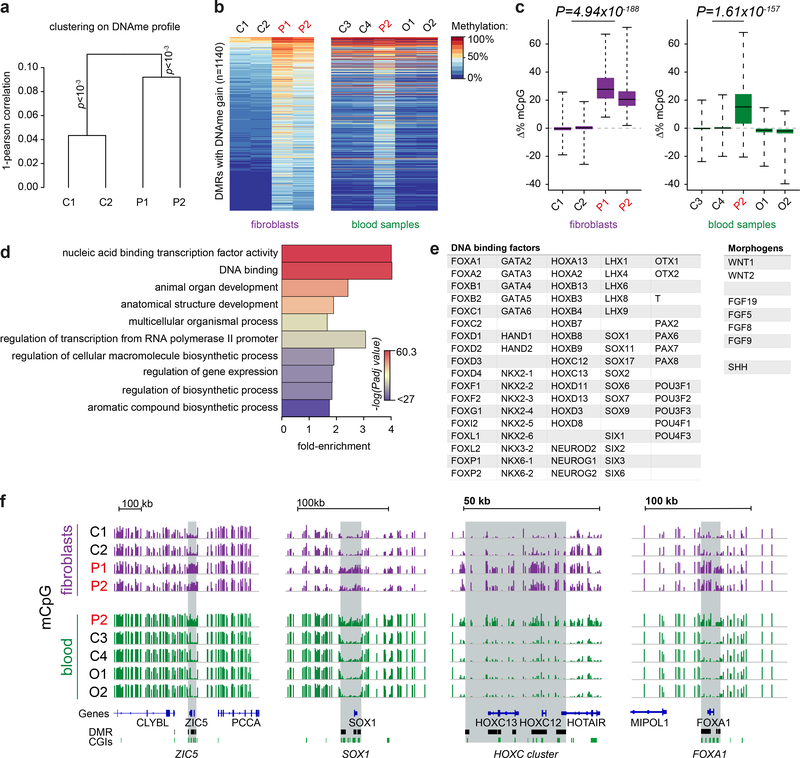
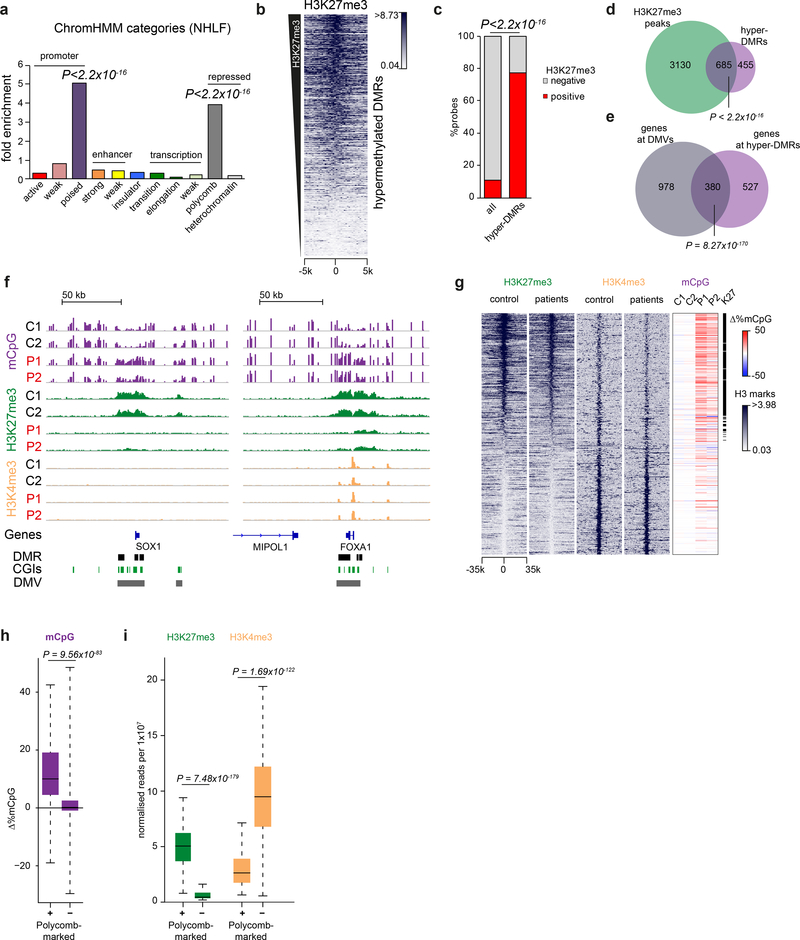
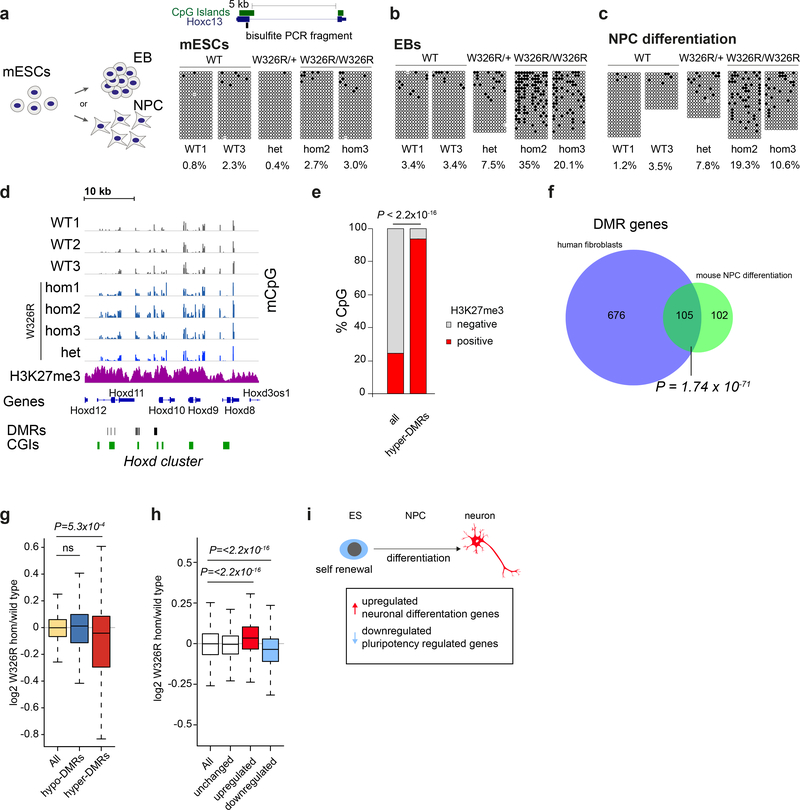
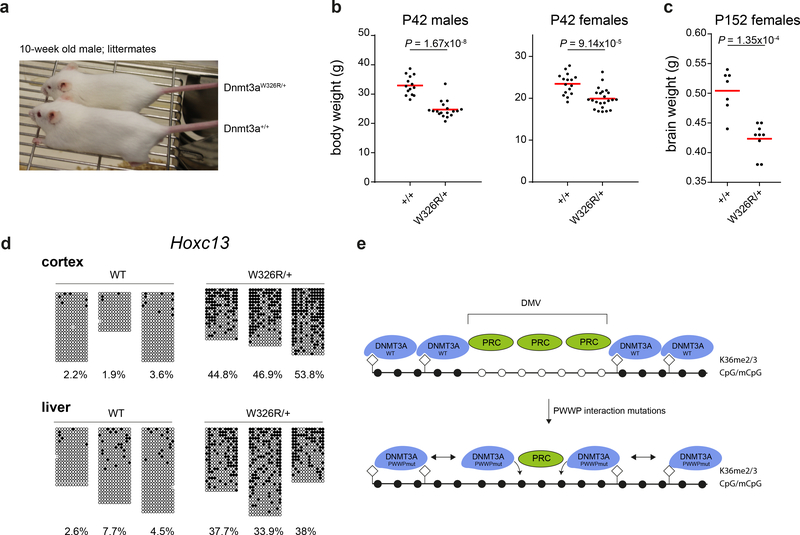
DNA methylation and Polycomb are key factors in the establishment of vertebrate cellular identity and fate. Here we report de novo missense mutations in DNMT3A, which encodes the DNA methyltransferase DNMT3A. These mutations cause microcephalic dwarfism, a hypocellular disorder of extreme global growth failure. Substitutions in the PWWP domain abrogate binding to the histone modifications H3K36me2 and H3K36me3, and alter DNA methylation in patient cells. Polycomb-associated DNA methylation valleys, hypomethylated domains encompassing developmental genes, become methylated with concomitant depletion of H3K27me3 and H3K4me3 bivalent marks. Such de novo DNA methylation occurs during differentiation of Dnmt3aW326R pluripotent cells in vitro, and is also evident in Dnmt3aW326R/+ dwarf mice. We therefore propose that the interaction of the DNMT3A PWWP domain with H3K36me2 and H3K36me3 normally limits DNA methylation of Polycomb-marked regions. Our findings implicate the interplay between DNA methylation and Polycomb at key developmental regulators as a determinant of organism size in mammals.
Conflict of interest statement
Competing Interests:
The authors declare no competing interests.
Figures
References
-
- Bicknell LS et al. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet 43, 350–5 (2011). - PubMed
-
- Guernsey DL et al. Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat Genet 43, 360–4 (2011). - PubMed
Methods only references:
-
- Barski A et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–37 (2007). - PubMed
-
- Guerois R, Nielsen JE & Serrano L Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 320, 369–87 (2002). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
