

Predicting the mutations generated by repair of Cas9-induced double-strand breaks
- PMID: 30480667
- PMCID: PMC6949135
- DOI: 10.1038/nbt.4317
Predicting the mutations generated by repair of Cas9-induced double-strand breaks
Abstract
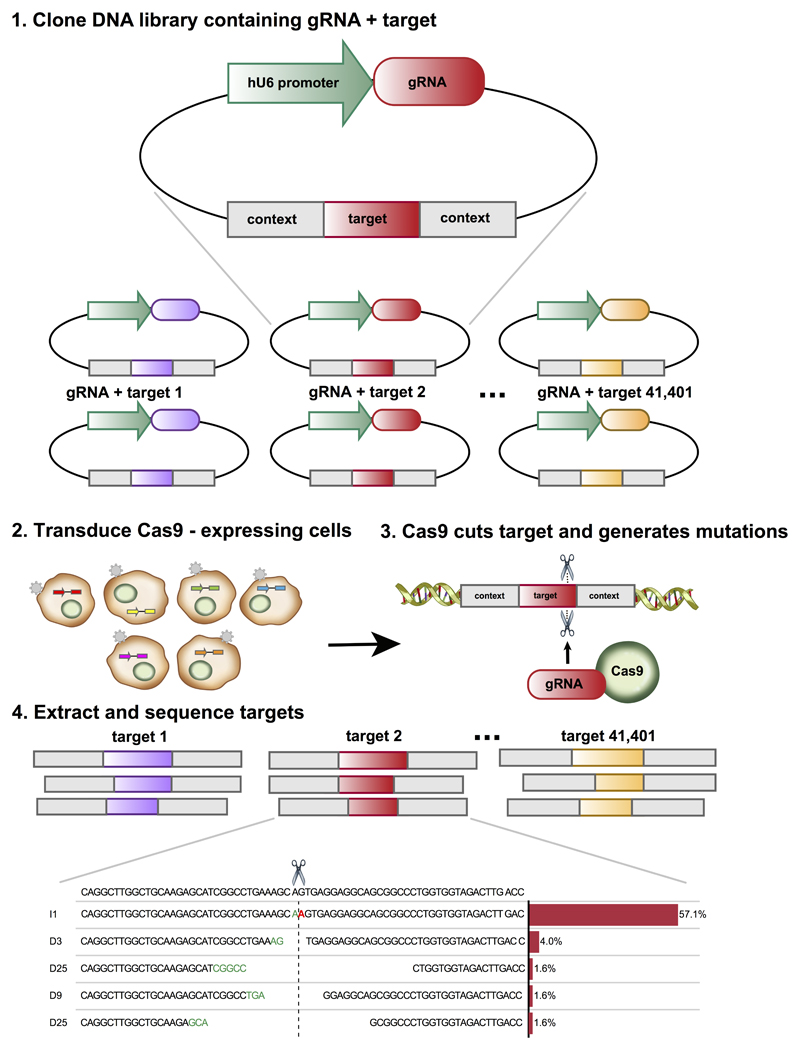
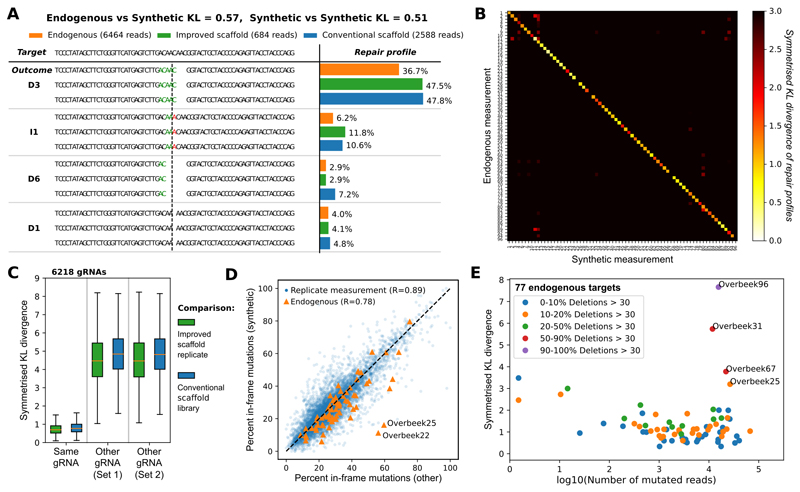
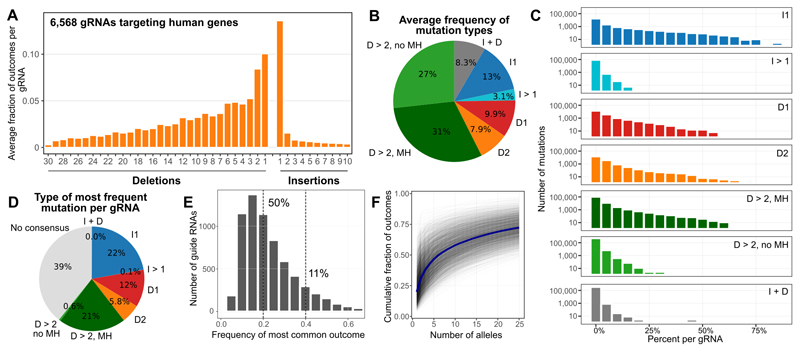
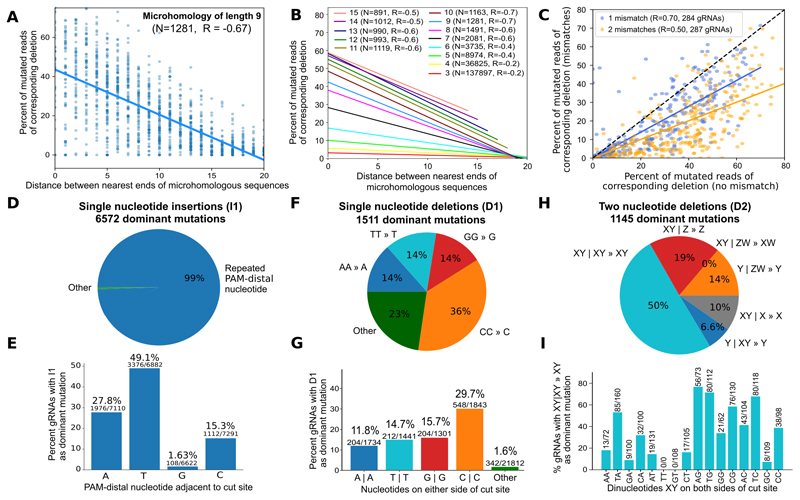
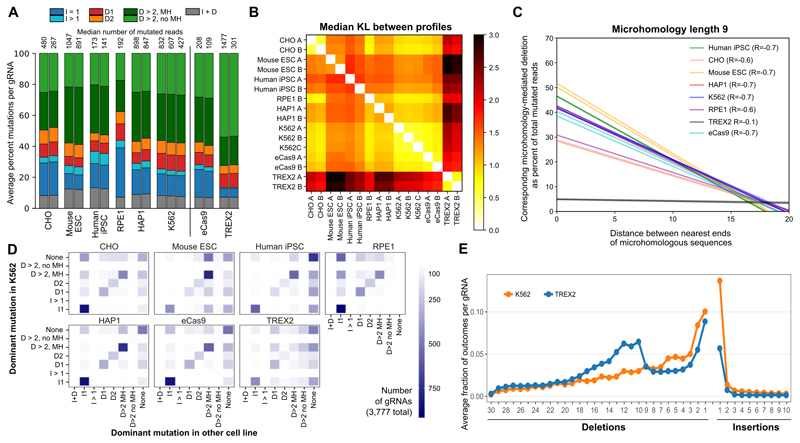
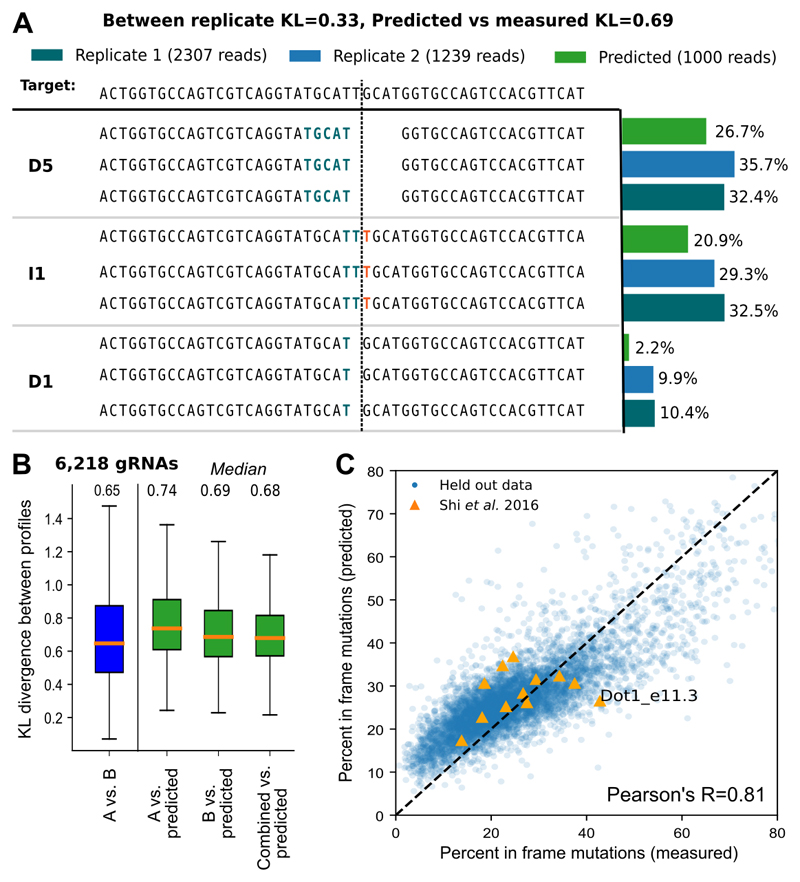
The DNA mutation produced by cellular repair of a CRISPR-Cas9-generated double-strand break determines its phenotypic effect. It is known that the mutational outcomes are not random, but depend on DNA sequence at the targeted location. Here we systematically study the influence of flanking DNA sequence on repair outcome by measuring the edits generated by >40,000 guide RNAs (gRNAs) in synthetic constructs. We performed the experiments in a range of genetic backgrounds and using alternative CRISPR-Cas9 reagents. In total, we gathered data for >109 mutational outcomes. The majority of reproducible mutations are insertions of a single base, short deletions or longer microhomology-mediated deletions. Each gRNA has an individual cell-line-dependent bias toward particular outcomes. We uncover sequence determinants of the mutations produced and use these to derive a predictor of Cas9 editing outcomes. Improved understanding of sequence repair will allow better design of gene editing experiments.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
