

The genome of the tegu lizard Salvator merianae: combining Illumina, PacBio, and optical mapping data to generate a highly contiguous assembly
- PMID: 30481296
- PMCID: PMC6304105
- DOI: 10.1093/gigascience/giy141
The genome of the tegu lizard Salvator merianae: combining Illumina, PacBio, and optical mapping data to generate a highly contiguous assembly
Abstract
Background: Reptiles are a species-rich group with great phenotypic and life history diversity but are highly underrepresented among the vertebrate species with sequenced genomes.
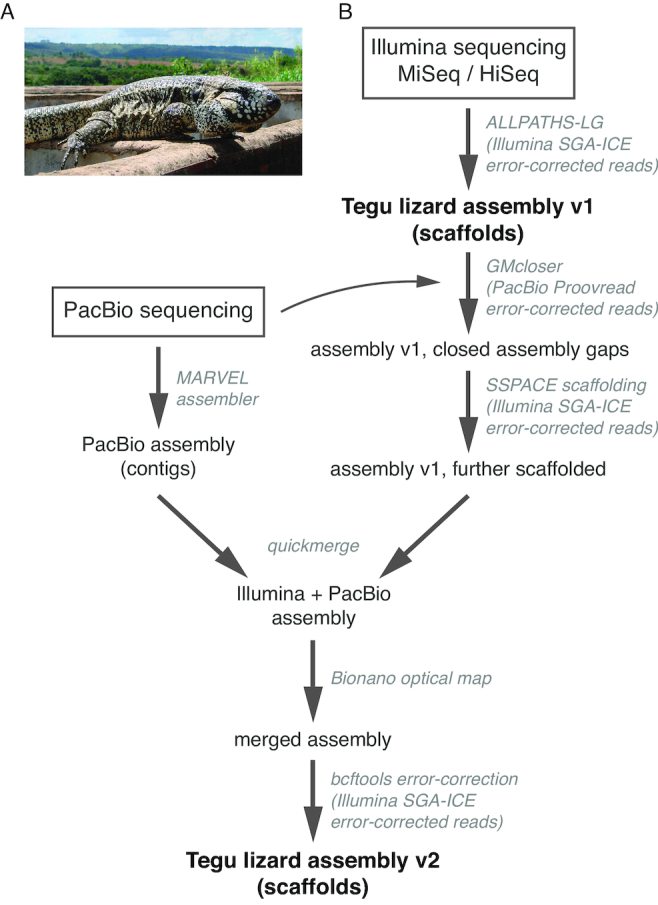
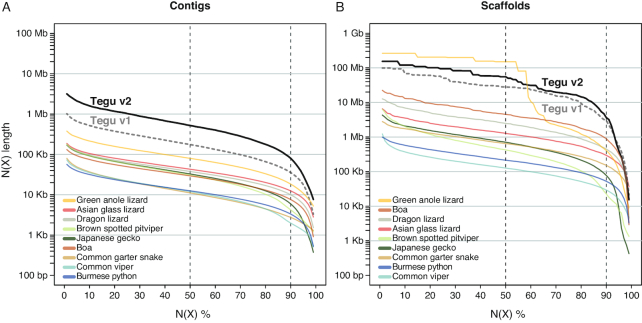
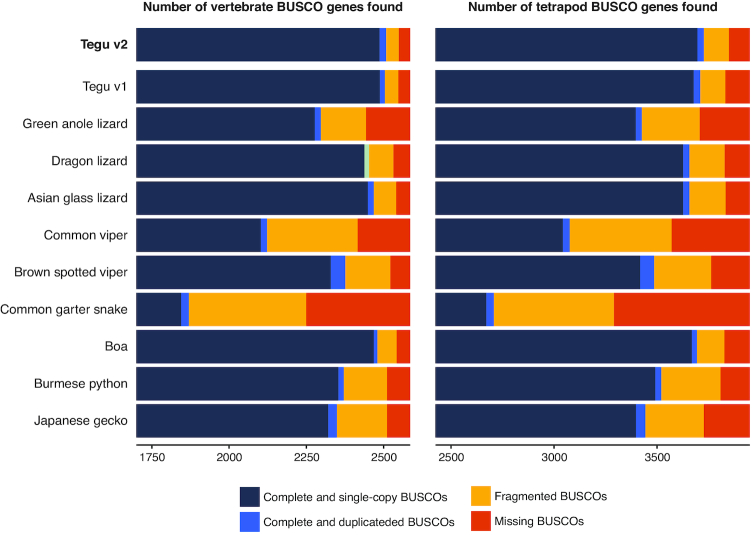
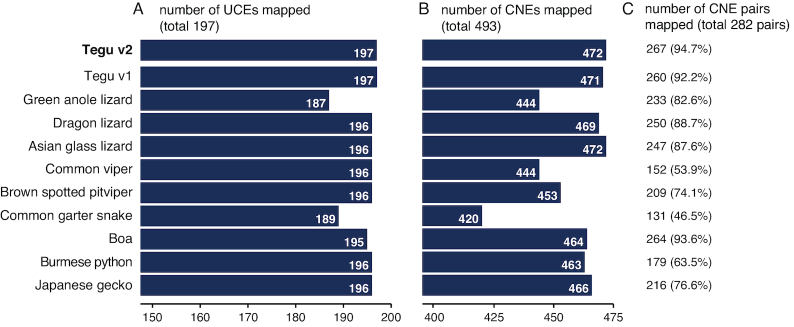
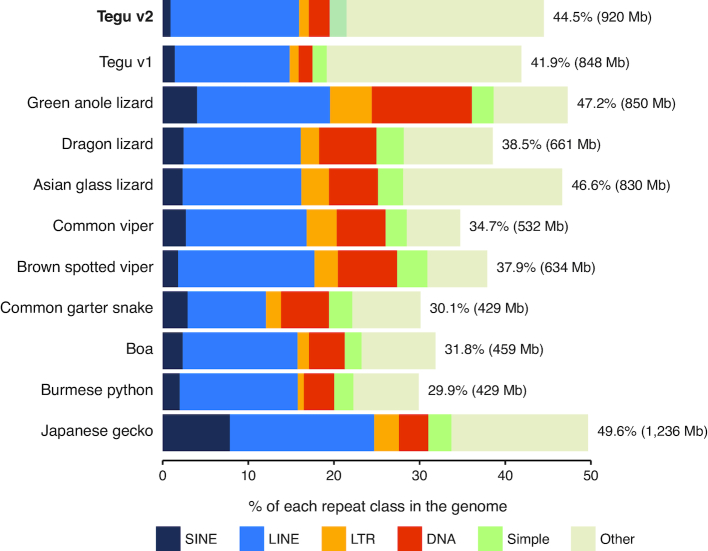
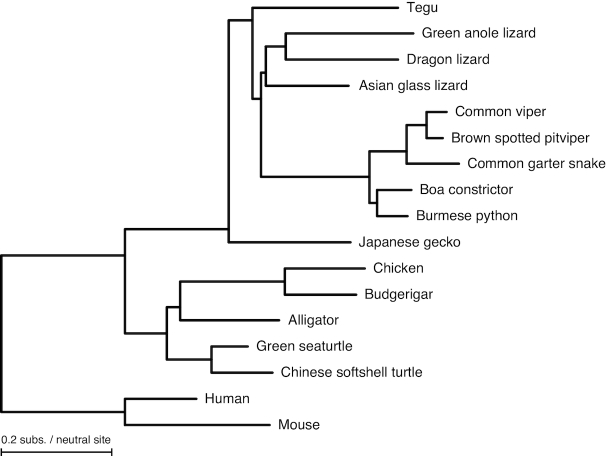
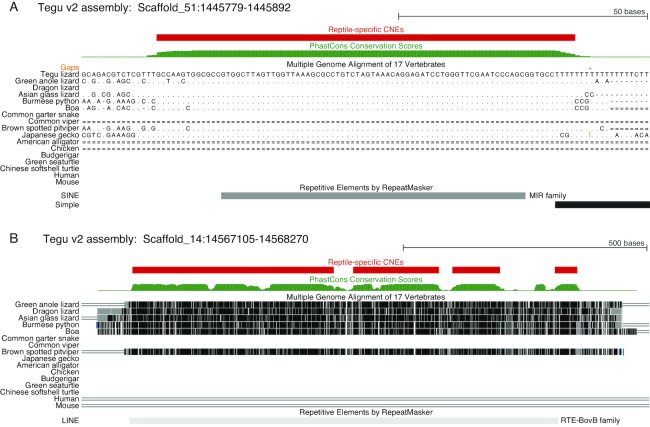
Results: Here, we report a high-quality genome assembly of the tegu lizard, Salvator merianae, the first lacertoid with a sequenced genome. We combined 74X Illumina short-read, 29.8X Pacific Biosciences long-read, and optical mapping data to generate a high-quality assembly with a scaffold N50 value of 55.4 Mb. The contig N50 value of this assembly is 521 Kb, making it the most contiguous reptile assembly so far. We show that the tegu assembly has the highest completeness of coding genes and conserved non-exonic elements (CNEs) compared to other reptiles. Furthermore, the tegu assembly has the highest number of evolutionarily conserved CNE pairs, corroborating a high assembly contiguity in intergenic regions. As in other reptiles, long interspersed nuclear elements comprise the most abundant transposon class. We used transcriptomic data, homology- and de novo gene predictions to annotate 22,413 coding genes, of which 16,995 (76%) likely have human orthologs as inferred by CESAR-derived gene mappings. Finally, we generated a multiple genome alignment comprising 10 squamates and 7 other amniote species and identified conserved regions that are under evolutionary constraint. CNEs cover 38 Mb (1.8%) of the tegu genome, with 3.3 Mb in these elements being squamate specific. In contrast to placental mammal-specific CNEs, very few of these squamate-specific CNEs (<20 Kb) overlap transposons, highlighting a difference in how lineage-specific CNEs originated in these two clades.
Conclusions: The tegu lizard genome together with the multiple genome alignment and comprehensive conserved element datasets provide a valuable resource for comparative genomic studies of reptiles and other amniotes.
Figures
Similar articles
-
Chromosome-Level Assembly of the Common Lizard (Zootoca vivipara) Genome.Genome Biol Evol. 2020 Nov 3;12(11):1953-1960. doi: 10.1093/gbe/evaa161. Genome Biol Evol. 2020. PMID: 32835354 Free PMC article.
-
Chromosome-Level Genome Assembly of the Cape Cliff Lizard (Hemicordylus capensis).Genome Biol Evol. 2023 Feb 3;15(2):evad001. doi: 10.1093/gbe/evad001. Genome Biol Evol. 2023. PMID: 36624992 Free PMC article.
-
A high-quality reference genome for the critically endangered Aeolian wall lizard, Podarcis raffonei.J Hered. 2023 May 25;114(3):279-285. doi: 10.1093/jhered/esad014. J Hered. 2023. PMID: 36866448
-
The State of Squamate Genomics: Past, Present, and Future of Genome Research in the Most Speciose Terrestrial Vertebrate Order.Genes (Basel). 2023 Jul 1;14(7):1387. doi: 10.3390/genes14071387. Genes (Basel). 2023. PMID: 37510292 Free PMC article. Review.
-
A lizard is never late: Squamate genomics as a recent catalyst for understanding sex chromosome and microchromosome evolution.J Hered. 2023 Aug 23;114(5):445-458. doi: 10.1093/jhered/esad023. J Hered. 2023. PMID: 37018459 Free PMC article.
Cited by
-
A chromosome-level genome assembly and annotation of the desert horned lizard, Phrynosoma platyrhinos, provides insight into chromosomal rearrangements among reptiles.Gigascience. 2022 Feb 4;11:giab098. doi: 10.1093/gigascience/giab098. Gigascience. 2022. PMID: 35134927 Free PMC article.
-
Comparative and evolutionary analysis of the reptilian hedgehog gene family (Shh, Dhh, and Ihh).PeerJ. 2019 Aug 30;7:e7613. doi: 10.7717/peerj.7613. eCollection 2019. PeerJ. 2019. PMID: 31531274 Free PMC article.
-
A chromosome-level genome assembly of an alpine plant Crucihimalaya lasiocarpa provides insights into high-altitude adaptation.DNA Res. 2022 Jan 28;29(1):dsac004. doi: 10.1093/dnares/dsac004. DNA Res. 2022. PMID: 35094078 Free PMC article.
-
Erythrocytes 3D genome organization in vertebrates.Sci Rep. 2021 Feb 24;11(1):4414. doi: 10.1038/s41598-021-83903-9. Sci Rep. 2021. PMID: 33627746 Free PMC article.
-
An updated reference genome of Barbatula barbatula (Linnaeus, 1758).Sci Data. 2025 Jan 22;12(1):137. doi: 10.1038/s41597-025-04469-z. Sci Data. 2025. PMID: 39843539 Free PMC article.
References
-
- Uetz P. The reptile database. http://www.reptile-database.org. Accessed March 2018.
-
- Crotalus genome. https://www.ncbi.nlm.nih.gov/assembly/727941. Accessed February 2018.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
