

A role for the unfolded protein response stress sensor ERN1 in regulating the response to MEK inhibitors in KRAS mutant colon cancers
- PMID: 30482246
- PMCID: PMC6258447
- DOI: 10.1186/s13073-018-0600-z
A role for the unfolded protein response stress sensor ERN1 in regulating the response to MEK inhibitors in KRAS mutant colon cancers
Erratum in
-
Correction to: A role for the unfolded protein response stress sensor ERN1 in regulating the response to MEK inhibitors in KRAS mutant colon cancers.Genome Med. 2021 Feb 10;13(1):24. doi: 10.1186/s13073-020-00815-5. Genome Med. 2021. PMID: 33568196 Free PMC article. No abstract available.
Abstract
Background: Mutations in KRAS are frequent in human cancer, yet effective targeted therapeutics for these cancers are still lacking. Attempts to drug the MEK kinases downstream of KRAS have had limited success in clinical trials. Understanding the specific genomic vulnerabilities of KRAS-driven cancers may uncover novel patient-tailored treatment options.
Methods: We first searched for synthetic lethal (SL) genetic interactions with mutant RAS in yeast with the ultimate aim to identify novel cancer-specific targets for therapy. Our method used selective ploidy ablation, which enables replication of cancer-specific gene expression changes in the yeast gene disruption library. Second, we used a genome-wide CRISPR/Cas9-based genetic screen in KRAS mutant human colon cancer cells to understand the mechanistic connection between the synthetic lethal interaction discovered in yeast and downstream RAS signaling in human cells.
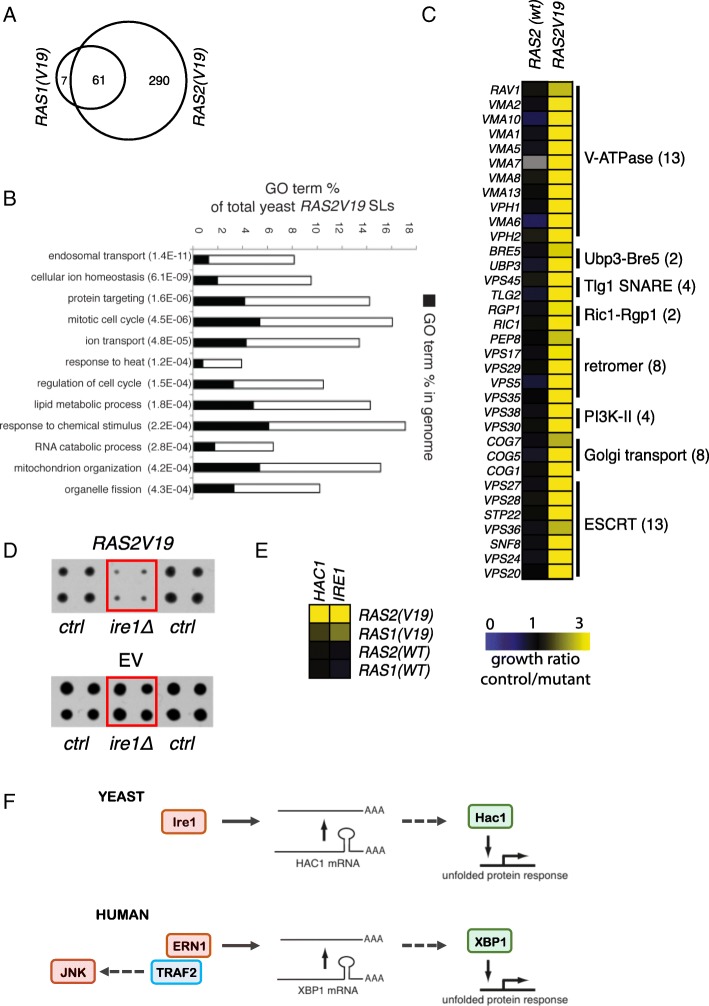
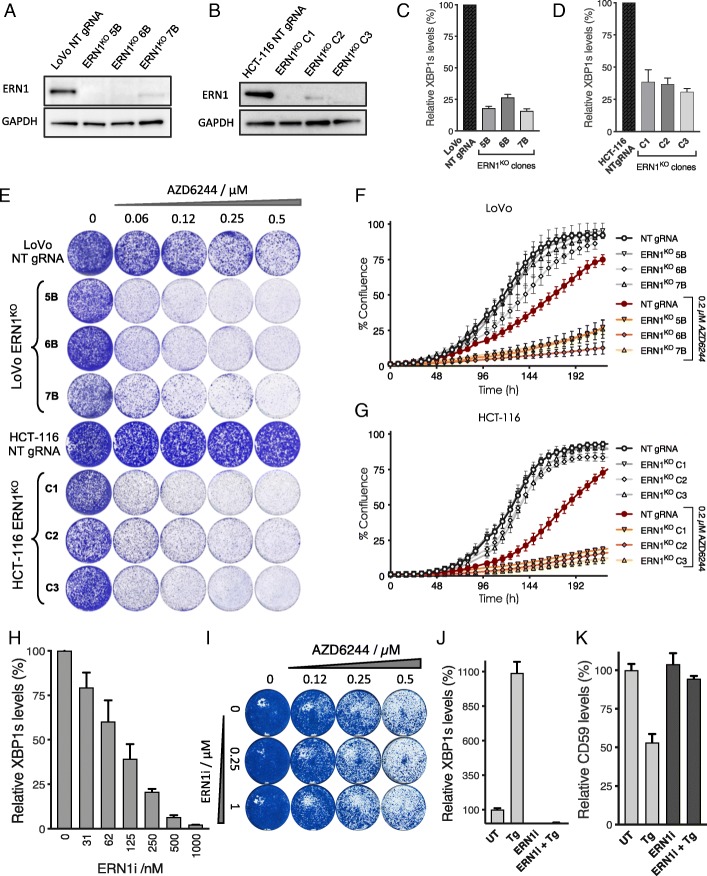
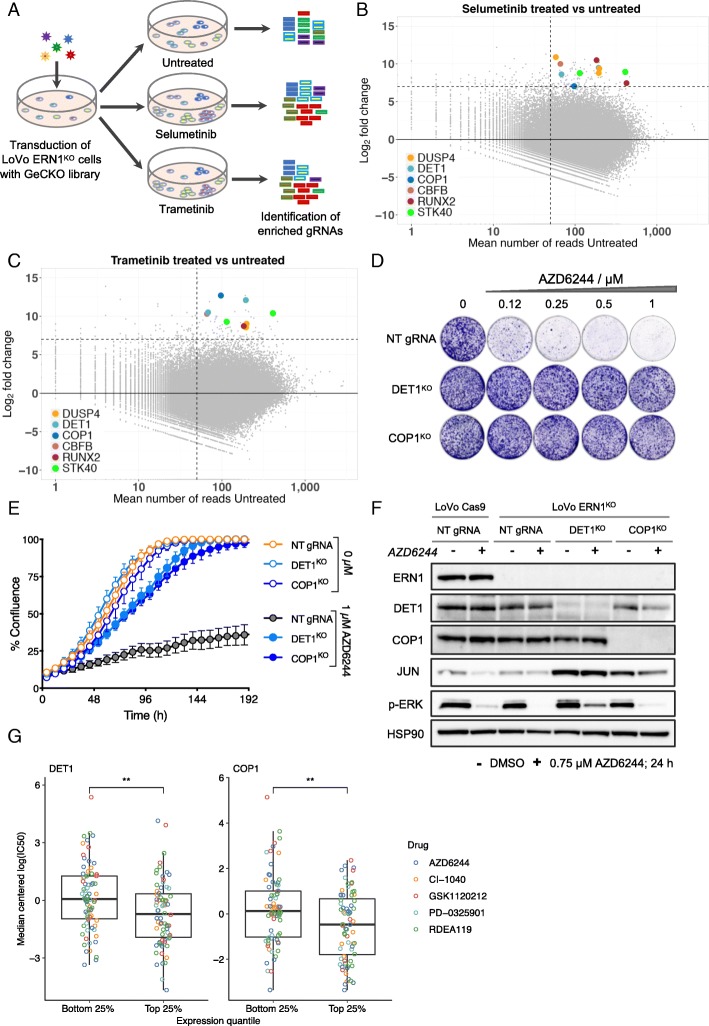
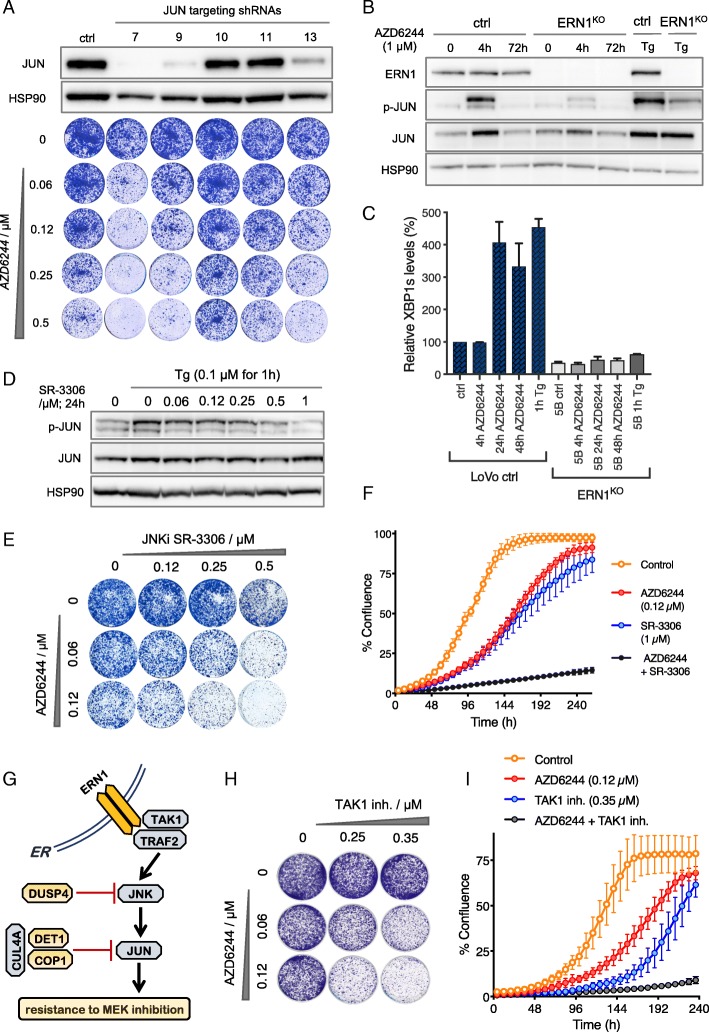
Results: We identify loss of the endoplasmic reticulum (ER) stress sensor IRE1 as synthetic lethal with activated RAS mutants in yeast. In KRAS mutant colorectal cancer cell lines, genetic ablation of the human ortholog of IRE1, ERN1, does not affect growth but sensitizes to MEK inhibition. However, an ERN1 kinase inhibitor failed to show synergy with MEK inhibition, suggesting that a non-kinase function of ERN1 confers MEK inhibitor resistance. To investigate how ERN1 modulates MEK inhibitor responses, we performed genetic screens in ERN1 knockout KRAS mutant colon cancer cells to identify genes whose inactivation confers resistance to MEK inhibition. This genetic screen identified multiple negative regulators of JUN N-terminal kinase (JNK) /JUN signaling. Consistently, compounds targeting JNK/MAPK8 or TAK1/MAP3K7, which relay signals from ERN1 to JUN, display synergy with MEK inhibition.
Conclusions: We identify the ERN1-JNK-JUN pathway as a novel regulator of MEK inhibitor response in KRAS mutant colon cancer. The notion that multiple signaling pathways can activate JUN may explain why KRAS mutant tumor cells are traditionally seen as highly refractory to MEK inhibitor therapy. Our findings emphasize the need for the development of new therapeutics targeting JUN activating kinases, TAK1 and JNK, to sensitize KRAS mutant cancer cells to MEK inhibitors.
Keywords: Colon cancer; ERN1; Ire1; JNK; JUN; MEK inhibitor.
Conflict of interest statement
Authors’ information
RLB is an associate professor at the Division of Molecular Carcinogenesis and the head of High Content Screening Facility at the Netherlands Cancer Institute. LW is a professor of Computational Cancer Biology at the Technical University in Delft and the co-director of the Cancer Systems Biology Center at the Netherlands Cancer Institute. RJDR is a Research Associate in the Department of Genetics & Development at Columbia University Vagelos College of Physicians & Surgeons. RR is a professor of Genetics & Development and Systems Biology at Columbia University Vagelos College of Physicians & Surgeons. RB is a professor of Molecular Carcinogenesis at the Netherlands Cancer Institute.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
MEK and TAK1 Regulate Apoptosis in Colon Cancer Cells with KRAS-Dependent Activation of Proinflammatory Signaling.Mol Cancer Res. 2016 Dec;14(12):1204-1216. doi: 10.1158/1541-7786.MCR-16-0173. Epub 2016 Sep 21. Mol Cancer Res. 2016. PMID: 27655129 Free PMC article.
-
Targeting β-catenin overcomes MEK inhibition resistance in colon cancer with KRAS and PIK3CA mutations.Br J Cancer. 2019 Apr;120(9):941-951. doi: 10.1038/s41416-019-0434-5. Epub 2019 Apr 4. Br J Cancer. 2019. PMID: 30944457 Free PMC article.
-
MEK nuclear localization promotes YAP stability via sequestering β-TrCP in KRAS mutant cancer cells.Cell Death Differ. 2019 Nov;26(11):2400-2415. doi: 10.1038/s41418-019-0309-6. Epub 2019 Mar 4. Cell Death Differ. 2019. PMID: 30833665 Free PMC article.
-
Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers.Int J Mol Sci. 2015 Sep 23;16(9):22976-88. doi: 10.3390/ijms160922976. Int J Mol Sci. 2015. PMID: 26404261 Free PMC article. Review.
-
Molecular pathways: the basis for rational combination using MEK inhibitors in KRAS-mutant cancers.Clin Cancer Res. 2014 Aug 15;20(16):4193-9. doi: 10.1158/1078-0432.CCR-13-2365. Epub 2014 Jun 6. Clin Cancer Res. 2014. PMID: 24907112 Review.
Cited by
-
Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer.Oncogene. 2022 Jan;41(2):191-203. doi: 10.1038/s41388-021-02077-w. Epub 2021 Oct 30. Oncogene. 2022. PMID: 34718347 Free PMC article.
-
From genome integrity to cancer.Genome Med. 2019 Jan 29;11(1):4. doi: 10.1186/s13073-019-0617-y. Genome Med. 2019. PMID: 30696459 Free PMC article. No abstract available.
-
Rad5 dysregulation drives hyperactive recombination at replication forks resulting in cisplatin sensitivity and genome instability.Nucleic Acids Res. 2019 Sep 26;47(17):9144-9159. doi: 10.1093/nar/gkz631. Nucleic Acids Res. 2019. PMID: 31350889 Free PMC article.
-
RUNX2/CBFB modulates the response to MEK inhibitors through activation of receptor tyrosine kinases in KRAS-mutant colorectal cancer.Transl Oncol. 2020 Feb;13(2):201-211. doi: 10.1016/j.tranon.2019.10.006. Epub 2019 Dec 20. Transl Oncol. 2020. PMID: 31865182 Free PMC article.
-
Evolution of Molecular Targeted Cancer Therapy: Mechanisms of Drug Resistance and Novel Opportunities Identified by CRISPR-Cas9 Screening.Front Oncol. 2022 Mar 17;12:755053. doi: 10.3389/fonc.2022.755053. eCollection 2022. Front Oncol. 2022. PMID: 35372044 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
