

Targeting BCL-xL improves the efficacy of bromodomain and extra-terminal protein inhibitors in triple-negative breast cancer by eliciting the death of senescent cells
- PMID: 30482844
- PMCID: PMC6341404
- DOI: 10.1074/jbc.RA118.004712
Targeting BCL-xL improves the efficacy of bromodomain and extra-terminal protein inhibitors in triple-negative breast cancer by eliciting the death of senescent cells
Abstract
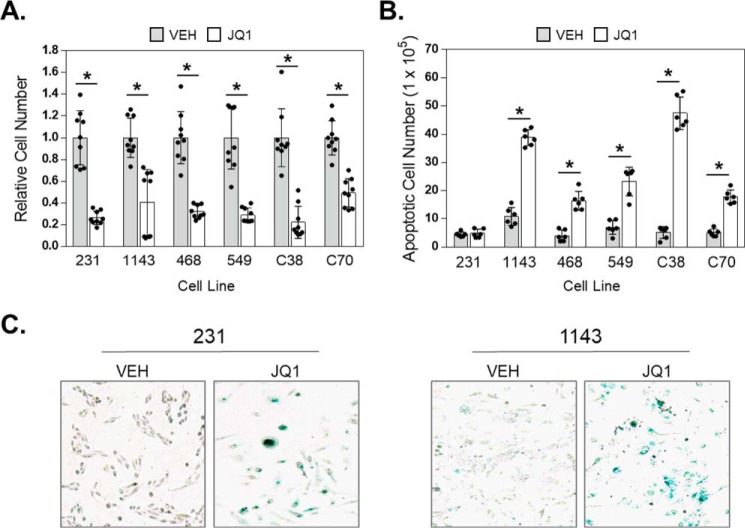
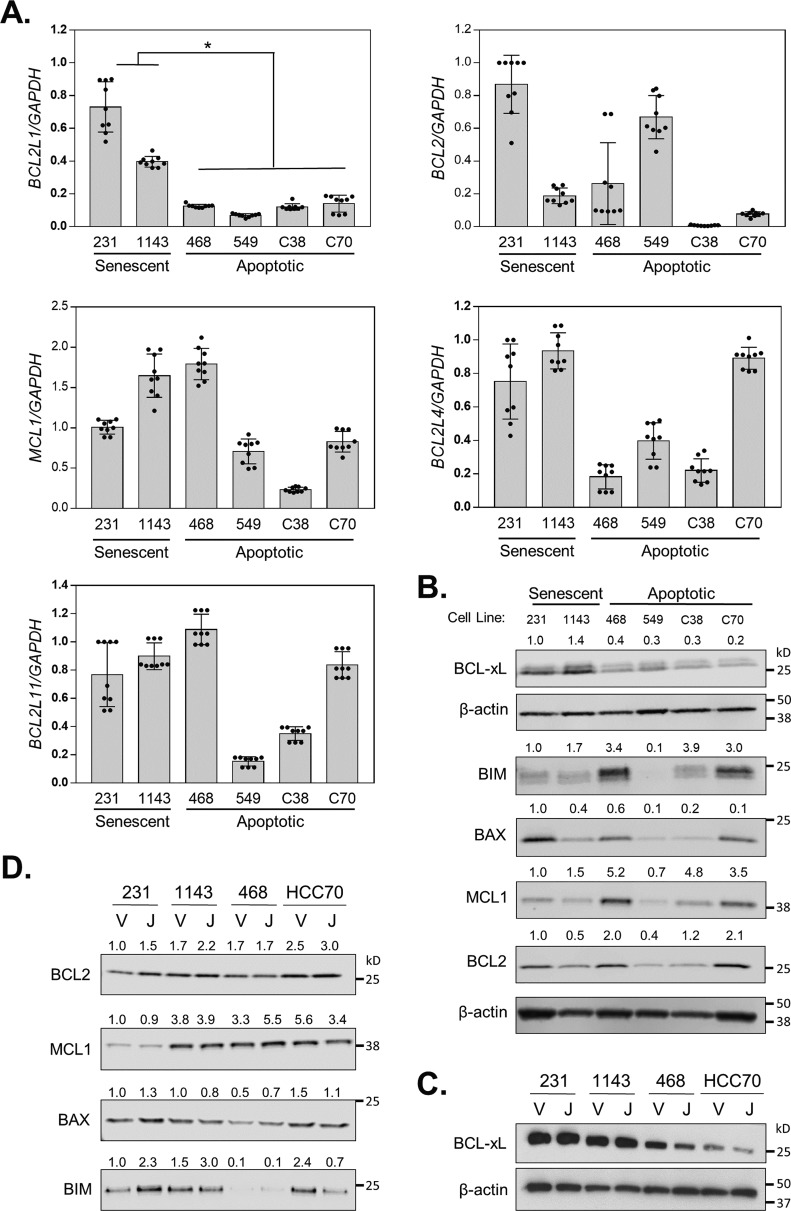
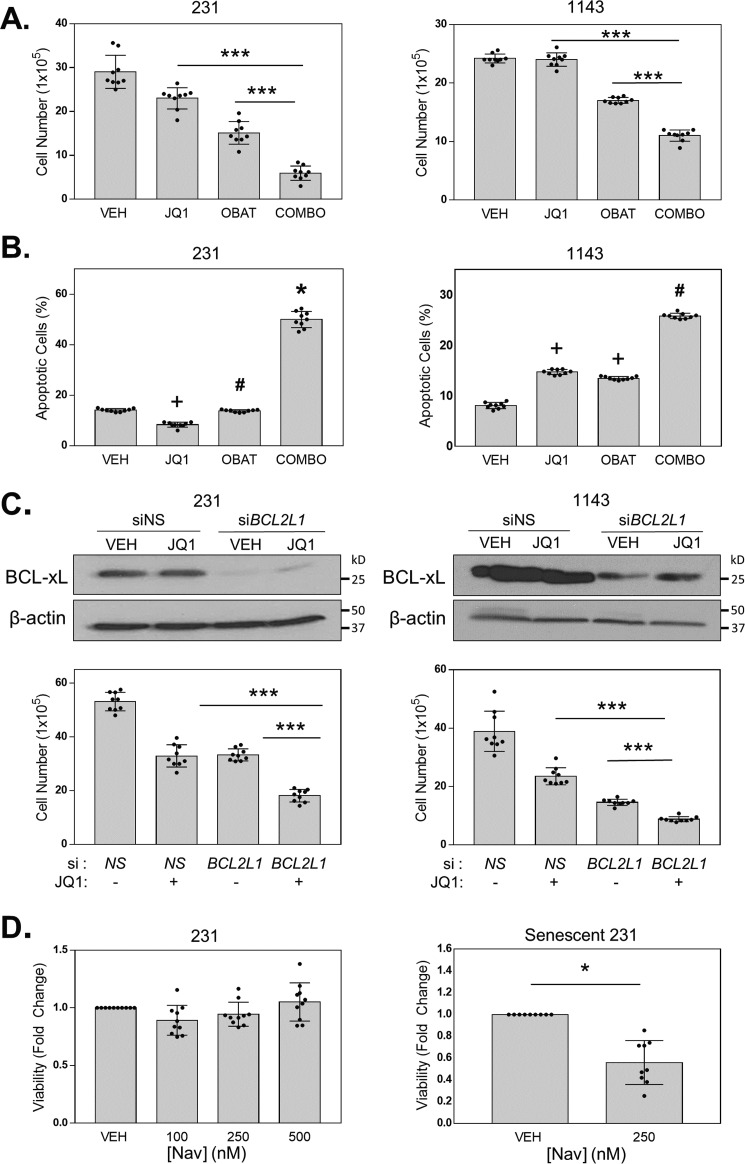
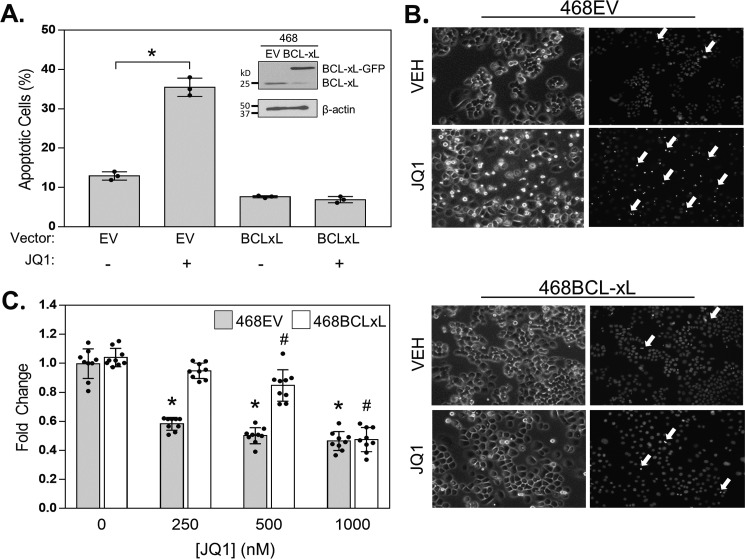
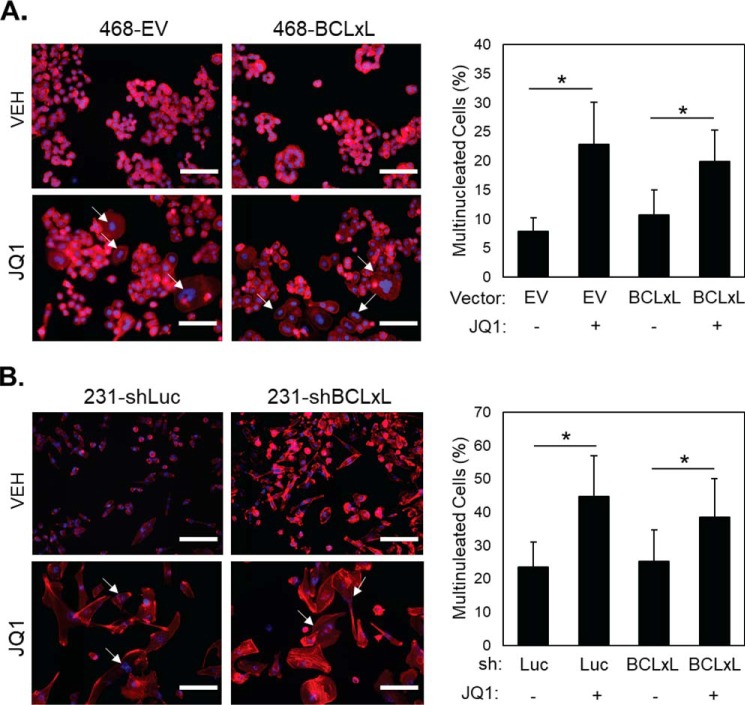
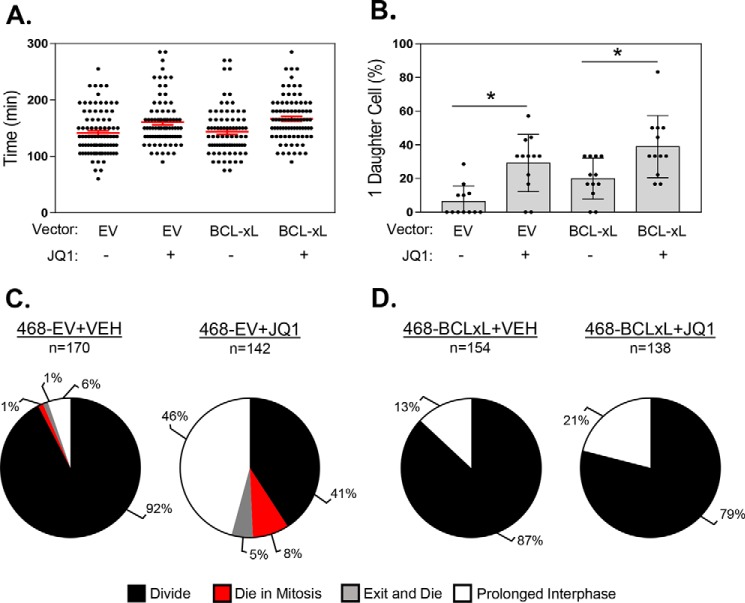
Inhibitors of bromodomain and extra-terminal proteins (BETi) suppress oncogenic gene expression and have been shown to be efficacious in many in vitro and murine models of cancer, including triple-negative breast cancer (TNBC), a highly aggressive disease. However, in most cancer models, responses to BETi can be highly variable. We previously reported that TNBC cells either undergo senescence or apoptosis in response to BETi, but the specific mechanisms dictating these two cell fates remain unknown. Using six human TNBC cell lines, we show that the terminal response of TNBC cells to BETi is dictated by the intrinsic expression levels of the anti-apoptotic protein B-cell lymphoma-extra large (BCL-xL). BCL-xL levels were higher in cell lines that senesce in response to BETi compared with lines that primarily die in response to these drugs. Moreover, BCL-xL expression was further reduced in cells that undergo BETi-mediated apoptosis. Forced BCL-xL overexpression in cells that normally undergo apoptosis following BETi treatment shifted them to senescence without affecting the reported mechanism of action of BETi in TNBC, that is, mitotic catastrophe. Most importantly, pharmacological or genetic inhibition of BCL-xL induced apoptosis in response to BETi, and inhibiting BCL-xL, even after BETi-induced senescence had already occurred, still induced cell death. These results indicate that BCL-xL provides a senescent cell death-inducing or senolytic target that may be exploited to improve therapeutic outcomes of TNBC in response to BETi. They also suggest that the basal levels of BCL-xL should be predictive of tumor responses to BETi in current clinical trials.
Keywords: B-cell lymphoma 2 (Bcl-2) family; B-cell lymphoma-extra large; BCL-xL; BCL2L1; BET inhibitor; apoptosis; breast cancer; bromodomain-containing protein 4 (BRD4); senescence; senolytic agent.
© 2019 Gayle et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
BCL-XL blockage in TNBC models confers vulnerability to inhibition of specific cell cycle regulators.Theranostics. 2021 Sep 3;11(19):9180-9197. doi: 10.7150/thno.60503. eCollection 2021. Theranostics. 2021. PMID: 34646365 Free PMC article.
-
Bromodomain and Extraterminal Protein Inhibition Blocks Growth of Triple-negative Breast Cancers through the Suppression of Aurora Kinases.J Biol Chem. 2016 Nov 4;291(45):23756-23768. doi: 10.1074/jbc.M116.738666. Epub 2016 Sep 20. J Biol Chem. 2016. PMID: 27650498 Free PMC article.
-
BET inhibitors (BETi) influence oxidative phosphorylation metabolism by affecting mitochondrial dynamics leading to alterations in apoptotic pathways in triple-negative breast cancer (TNBC) cells.Cell Prolif. 2024 Dec;57(12):e13730. doi: 10.1111/cpr.13730. Epub 2024 Sep 2. Cell Prolif. 2024. PMID: 39223828 Free PMC article.
-
Bcl-xL as a Modulator of Senescence and Aging.Int J Mol Sci. 2021 Feb 3;22(4):1527. doi: 10.3390/ijms22041527. Int J Mol Sci. 2021. PMID: 33546395 Free PMC article. Review.
-
The interplay between apoptosis and cellular senescence: Bcl-2 family proteins as targets for cancer therapy.Pharmacol Ther. 2022 Feb;230:107943. doi: 10.1016/j.pharmthera.2021.107943. Epub 2021 Jun 25. Pharmacol Ther. 2022. PMID: 34182005 Review.
Cited by
-
BCL-XL blockage in TNBC models confers vulnerability to inhibition of specific cell cycle regulators.Theranostics. 2021 Sep 3;11(19):9180-9197. doi: 10.7150/thno.60503. eCollection 2021. Theranostics. 2021. PMID: 34646365 Free PMC article.
-
BCL-xL, a Mitochondrial Protein Involved in Successful Aging: From C. elegans to Human Centenarians.Int J Mol Sci. 2020 Jan 9;21(2):418. doi: 10.3390/ijms21020418. Int J Mol Sci. 2020. PMID: 31936510 Free PMC article. Review.
-
Modulation of tumor plasticity by senescent cells: Deciphering basic mechanisms and survival pathways to unravel therapeutic options.Genet Mol Biol. 2024 May 27;47Suppl 1(Suppl 1):e20230311. doi: 10.1590/1678-4685-GMB-2023-0311. eCollection 2024. Genet Mol Biol. 2024. PMID: 38805699 Free PMC article.
-
Aurora Kinase A and Bcl-xL Inhibition Suppresses Metastasis in Triple-Negative Breast Cancer.Int J Mol Sci. 2022 Sep 2;23(17):10053. doi: 10.3390/ijms231710053. Int J Mol Sci. 2022. PMID: 36077449 Free PMC article.
-
Could senescence phenotypes strike the balance to promote tumor dormancy?Cancer Metastasis Rev. 2023 Mar;42(1):143-160. doi: 10.1007/s10555-023-10089-z. Epub 2023 Feb 3. Cancer Metastasis Rev. 2023. PMID: 36735097 Free PMC article. Review.
References
-
- Carey L. A., Perou C. M., Livasy C. A., Dressler L. G., Cowan D., Conway K., Karaca G., Troester M. A., Tse C. K., Edmiston S., Deming S. L., Geradts J., Cheang M. C., Nielsen T. O., Moorman P. G., Earp H. S., and Millikan R. C. (2006) Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 295, 2492–2502 10.1074/jbc.RA117.001068 - DOI - PubMed
-
- Badve S., Dabbs D. J., Schnitt S. J., Baehner F. L., Decker T., Eusebi V., Fox S. B., Ichihara S., Jacquemier J., Lakhani S. R., Palacios J., Rakha E. A., Richardson A. L., Schmitt F. C., Tan P. H., Tse G. M., Weigelt B., Ellis I. O., and Reis-Filho J. S. (2011) Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 24, 157–167 10.1074/jbc.RA117.001068 - DOI - PubMed
-
- Bauer K. R., Brown M., Cress R. D., Parise C. A., and Caggiano V. (2007) Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California Cancer Registry. Cancer 109, 1721–1728 10.1074/jbc.RA117.001068 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
